
A Comparative Analysis of Bacterial and Fungal Communities in Coastal and Inland Pecan Plantations

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Site and Soil Sample Collection
2.2. Determination of Soil Physicochemical Properties
2.3. DNA Extraction and Polymerase Chain Reaction (PCR) Amplification
2.4. Illumina Sequencing
2.5. Sequence Data Processing
2.6. Bioinformatics Analysis
2.6.1. Community Composition Analysis
2.6.2. Indicator Species Analysis
2.6.3. Alpha Diversity Analysis
2.6.4. Beta Diversity Analysis
2.6.5. Environmental Factor and Network Analysis
2.6.6. Kyoto Encyclopedia of Genes and Genomes (KEGG) Functional Analysis
3. Results
3.1. Site Characteristics
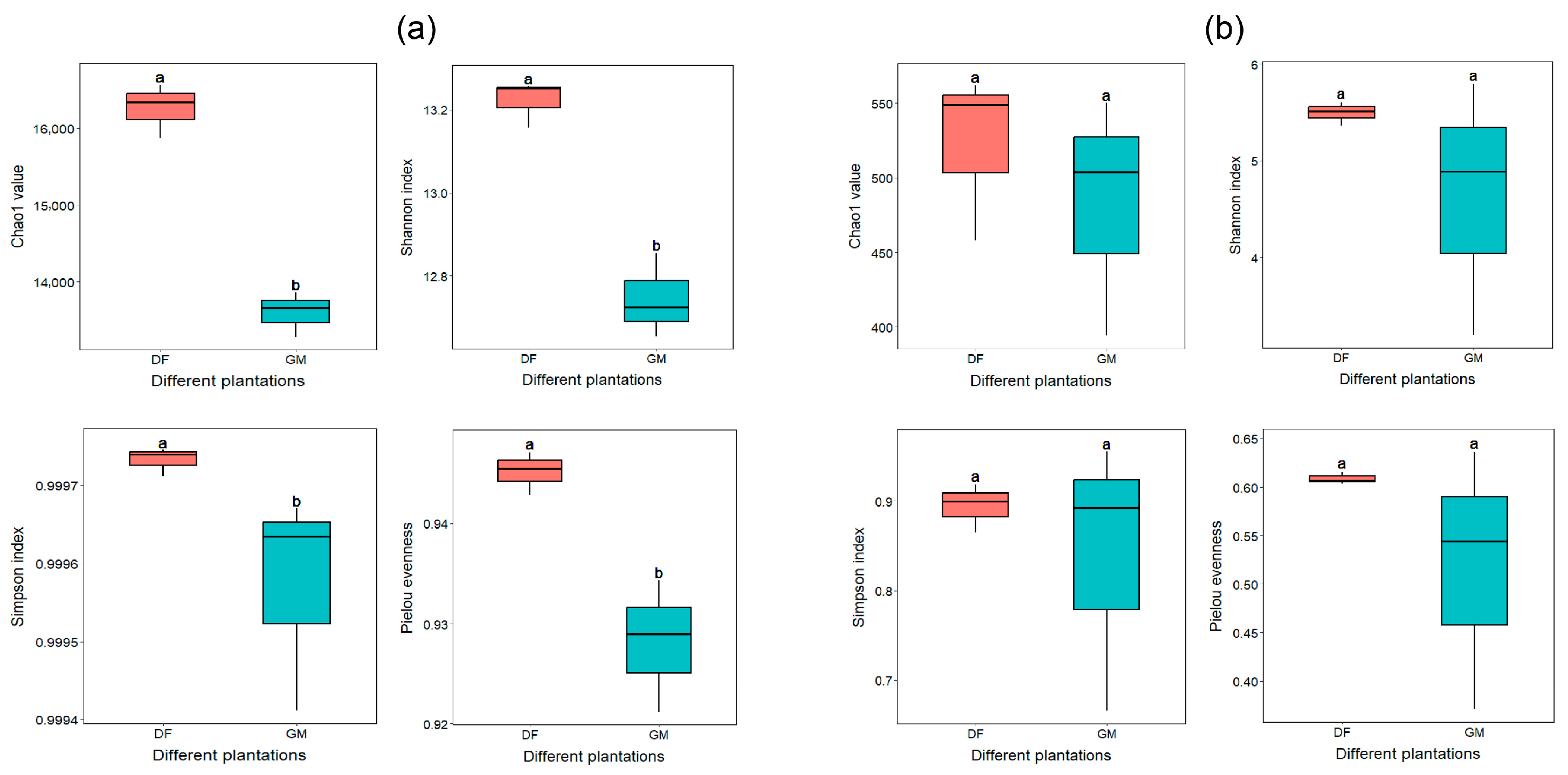
3.2. Soil Bacterial and Fungal Community Diversity and Structure
3.3. Soil Bacterial and Fungal Community Composition
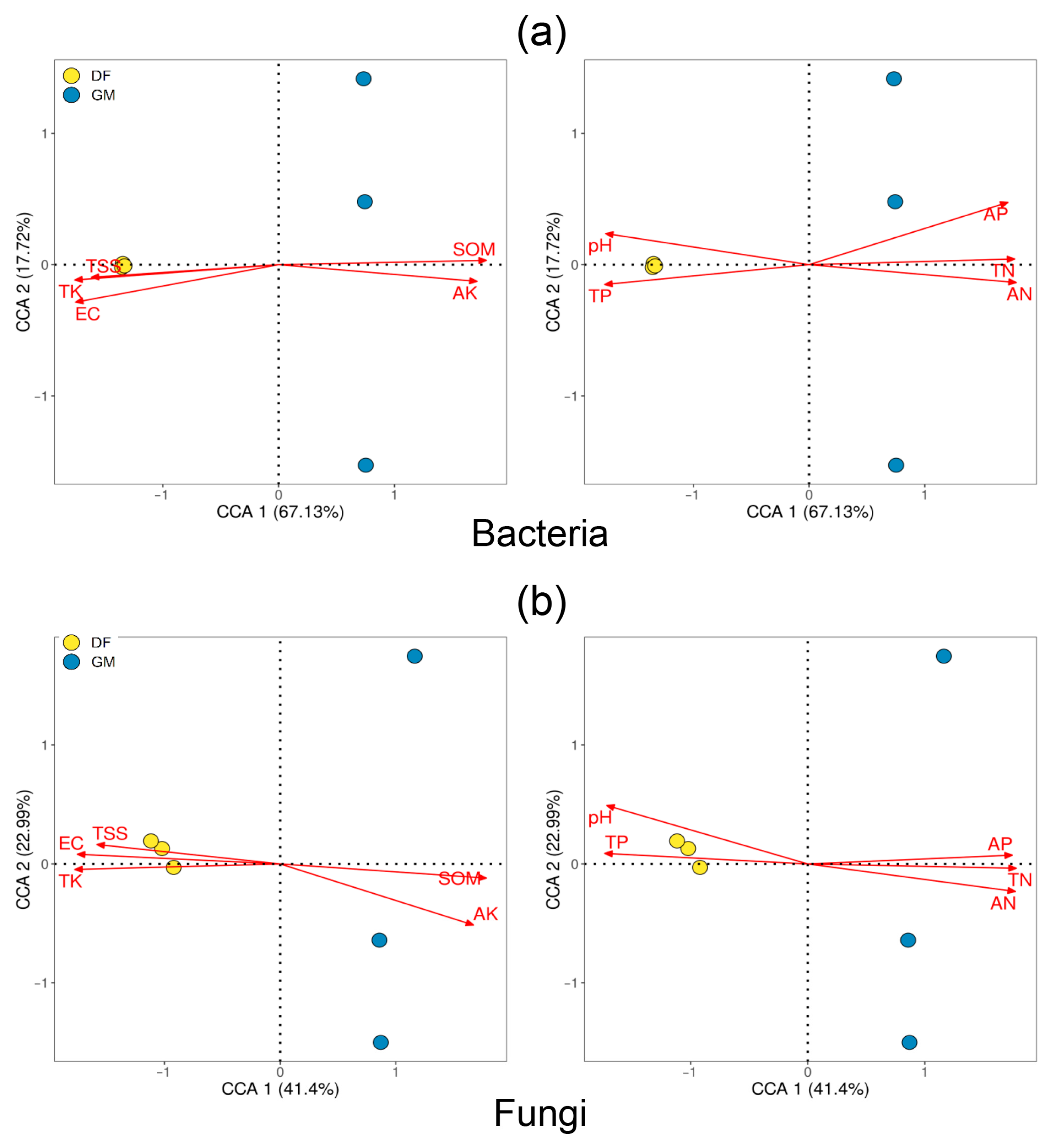
3.4. Differences in Soil Bacterial and Fungal Community Structure
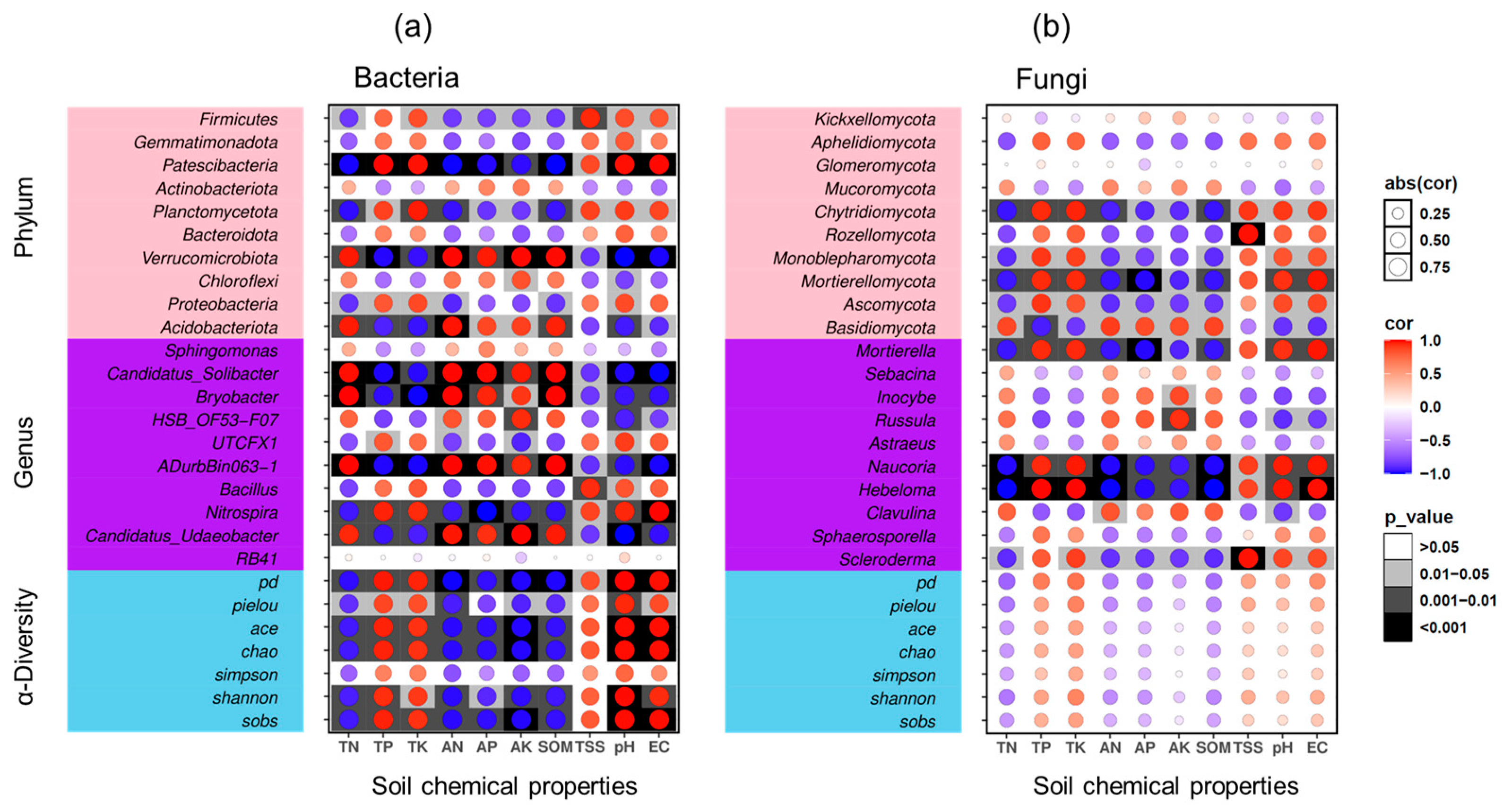
3.5. Correlation Analysis of Soil Chemical Properties and Microorganisms
3.6. Soil Microorganism Correlation Network Structures
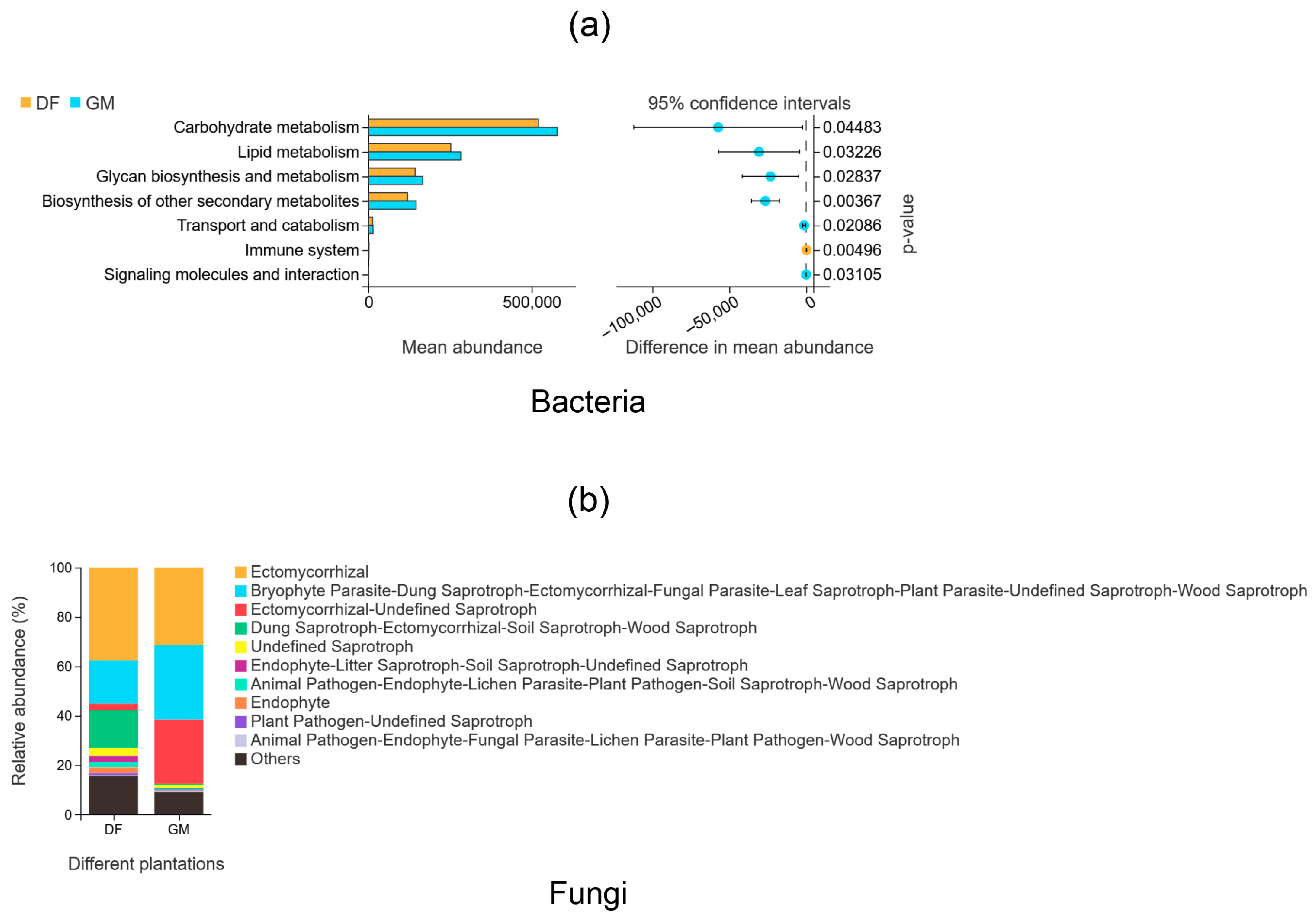
3.7. Functional Analyses of Soil Bacteria and Fungi
4. Discussion
4.1. Variation in Microbial Diversity between Coastal and Inland Pecan Plantations
4.2. Taxonomic Composition and Structural Changes in Soil Microbial Communities of Pecan Plantations
4.3. Relationships between Soil Chemical Properties and Microbial Community Structure
4.4. Soil Microbial Network Structures and Functions in Pecan Plantations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, Y.; Liu, J.; Bao, J.; Chu, G.; Peng, F. Soil type influences rhizosphere bacterial community assemblies of pecan plantations, a case study of eastern China. Forests 2022, 13, 363. [Google Scholar] [CrossRef]
- Wang, M.; Mo, Z.; Lin, R.; Zhu, C. Characterization and expression analysis of the SPL gene family during floral development and abiotic stress in pecan (Carya illinoinensis). PeerJ 2021, 9, e12490. [Google Scholar] [CrossRef]
- Manchester, S.R. Fossil History of the Juglandaceae. Ph.D. Dissertation, Indiana University, Bloomington, IN, USA, 1981. [Google Scholar]
- Cervantes, K.; Velasco-Cruz, C.; Grauke, L.; Wang, X.; Conner, P.; Wells, L.; Bock, C.H.; Pisani, C.; Randall, J.J. Influence of Geographical Orchard Location on the Microbiome from the Progeny of a Pecan Controlled Cross. Plants 2023, 12, 360. [Google Scholar] [CrossRef]
- Zhang, R.; Peng, F.; Li, Y. Pecan production in China. Sci. Hortic. 2015, 197, 719–727. [Google Scholar] [CrossRef]
- Liu, J.; Tang, Y.; Bao, J.; Wang, H.; Peng, F.; Chen, M.; Tan, P. Pecan plantation age influences the structures, ecological networks, and functions of soil microbial communities. Land Degrad. Dev. 2022, 33, 3294–3309. [Google Scholar] [CrossRef]
- Ramakuwela, T.; Hatting, J.; Bock, C.; Vega, F.E.; Wells, L.; Mbata, G.N.; Shapiro-Ilan, D. Establishment of Beauveria bassiana as a fungal endophyte in pecan (Carya illinoinensis) seedlings and its virulence against pecan insect pests. Biol. Control 2020, 140, 104102. [Google Scholar] [CrossRef]
- Zhu, K.; Fan, P.; Mo, Z.; Tan, P.; Feng, G.; Li, F.; Peng, F. Identification, expression and co-expression analysis of R2R3-MYB family genes involved in graft union formation in pecan (Carya illinoinensis). Forests 2020, 11, 917. [Google Scholar] [CrossRef]
- Chen, T.; Hu, R.; Zheng, Z.; Yang, J.; Fan, H.; Deng, X.; Yao, W.; Wang, Q.; Peng, S.; Li, J. Soil bacterial community in the multiple cropping system increased grain yield within 40 cultivation years. Front. Plant Sci. 2021, 12, 804527. [Google Scholar] [CrossRef]
- Xu, H.; Yu, M.; Cheng, X. Abundant fungal and rare bacterial taxa jointly reveal soil nutrient cycling and multifunctionality in uneven-aged mixed plantations. Ecol. Indic. 2021, 129, 107932. [Google Scholar] [CrossRef]
- Cervantes, K.; Heerema, R.J.; Randall, J.J. The core microbiome of Carya illinoinensis (pecan) seedlings of different maternal pecan cultivars from the same orchard. Front. Microbiomes 2022, 1, 1003112. [Google Scholar] [CrossRef]
- Langella, F.; Grawunder, A.; Stark, R.; Weist, A.; Merten, D.; Haferburg, G.; Büchel, G.; Kothe, E. Microbially assisted phytoremediation approaches for two multi-element contaminated sites. Environ. Sci. Pollut. Res. Int. 2014, 21, 6845–6858. [Google Scholar] [CrossRef]
- Liu, X.; Du, F.; Chen, S.; Li, N.; Cui, J.; Chang, Y.; Sun, L.; Li, J.; Yao, D. Increased diversity of rhizosphere bacterial community confers adaptability to coastal environment for Sapium sebiferum trees. Forests 2022, 13, 667. [Google Scholar] [CrossRef]
- Grauke, L. Pecan seed stock selection—Regional implications. Proc. SE Pecan Grow. Assoc. 2010, 103, 42–50. [Google Scholar]
- Cervantes, K. Microbiome Composition of Carya illinoinensis (pecan) Seedlings and the Effect of Deficit Watering on Micropropagated Pecan Trees and Their Associated Fungal Communities. Ph.D. Dissertation, New Mexico State University, Las Cruces, NM, USA, 2022. [Google Scholar]
- Yuan, Z. Infection of AMF of Main Afforestation Tree Species in Coastal Areas of Northern Jiangsu and Its Correlation with Edaphic Factors. Master’s Thesis, Nanjing Forestry University, Nanjing, China, 2019. [Google Scholar]
- Wen, Z.; Lin, C.; Xu, X.; Ma, S.; Peng, Y.; Sun, Y.; Tang, B.; Shi, L. Ectomycorrhizal community associated with Cedrus deodara in four urban forests of Nantong in East China. Front. Plant Sci. 2023, 14, 1226720. [Google Scholar] [CrossRef]
- Bao, S. Soil Agrochemical Analysis (for Professionals in Soil Science, Agricultural Chemistry, Resources, and Environmental Studies), 3rd ed.; China Agriculture Press: Beijing, China, 2008. [Google Scholar]
- Liu, X.; Lu, X.; Zhao, W.; Yang, S.; Wang, J.; Xia, H.; Wei, X.; Zhang, J.; Chen, L.; Chen, Q. The rhizosphere effect of native legume Albizzia julibrissin on coastal saline soil nutrient availability, microbial modulation, and aggregate formation. Sci. Total Environ. 2022, 806, 150705. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Suo, M.; Qiu, Z.; Wu, H.; Zhao, M.; Yang, H. Regulating root fungal community using Mortierella alpina for Fusarium oxysporum resistance in Panax ginseng. Front. Microbiol. 2022, 13, 850917. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, X.; Jiang, X.; Ding, B.; Wu, P.; Ding, F. Plant functional trait responses to dolomite and limestone karst forests in southwest china. Forests 2022, 13, 2187. [Google Scholar] [CrossRef]
- Guo, M.; Wu, F.; Hao, G.; Qi, Q.; Li, R.; Li, N.; Wei, L.; Chai, T. Bacillus subtilis improves immunity and disease resistance in rabbits. Front. Immunol. 2017, 8, 354–366. [Google Scholar] [CrossRef]
- Wang, P.; Ding, L.; Zou, C.; Zhang, Y.; Wang, M. Rhizosphere element circling, multifunctionality, aboveground productivity and trade-offs are better predicted by rhizosphere rare taxa. Front. Plant Sci. 2022, 13, 985574. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 1–10. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. WIRES Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1, 790. [Google Scholar]
- Revelle, W.; Revelle, M.W. Package ‘psych’. Compr. R Arch. Netw. 2015, 337, 161–165. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’Hara, R.B.; Simpson, G.; Solymos, P.; Henry, M.; Stevens, W. Vegan: Community Ecology Package. R Package, Version 1.17-4. 2010. Available online: https://cran.r-project.org (accessed on 1 November 2023).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Prober, S.M.; Leff, J.W.; Bates, S.T.; Borer, E.T.; Firn, J.; Harpole, W.S.; Lind, E.M.; Seabloom, E.W.; Adler, P.B.; Bakker, J.D. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol. Lett. 2015, 18, 85–95. [Google Scholar] [CrossRef]
- Ma, B.; Gong, J. A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils. World J. Microb. Biot. 2013, 29, 2325–2334. [Google Scholar] [CrossRef]
- Ren, W.; Zhang, L.; Maness, N.; Wang, X.; Tang, M.; Xu, T. Changes in the diversity of pecan (Carya illinoinensis) rhizosphere microbial community with different nitrogen fertilization, a case study in Oklahoma pecan orchard. Sci. Hortic. 2023, 321, 112365. [Google Scholar] [CrossRef]
- Otero-Jiménez, V.; del Pilar Carreno-Carreno, J.; Barreto-Hernandez, E.; van Elsas, J.D.; Uribe-Velez, D. Impact of rice straw management strategies on rice rhizosphere microbiomes. Appl. Soil Ecol. 2021, 167, 104036. [Google Scholar] [CrossRef]
- Zhang, H.; Ullah, F.; Ahmad, R.; Shah, S.U.A.; Khan, A.; Adnan, M. Response of soil proteobacteria to biochar amendment in sustainable agriculture-a mini review. J. Soil Plant Environ. 2022, 1, 16–30. [Google Scholar] [CrossRef]
- Mhete, M.; Eze, P.N.; Rahube, T.O.; Akinyemi, F.O. Soil properties influence bacterial abundance and diversity under different land-use regimes in semi-arid environments. Sci. Afr. 2020, 7, e00246. [Google Scholar] [CrossRef]
- Li, M.; Chen, Z.; Qian, J.; Wei, F.; Zhang, G.; Wang, Y.; Wei, G.; Hu, Z.; Dong, L.; Chen, S. Composition and function of rhizosphere microbiome of Panax notoginseng with discrepant yields. Chin. Med. 2020, 15, 85. [Google Scholar] [CrossRef]
- Willms, I.M.; Bolz, S.H.; Yuan, J.; Krafft, L.; Schneider, D.; Schöning, I.; Schrumpf, M.; Nacke, H. The ubiquitous soil verrucomicrobial clade ‘Candidatus Udaeobacter’shows preferences for acidic pH. Environ. Microbiol. Rep. 2021, 13, 878–883. [Google Scholar] [CrossRef]
- Ercole, T.G.; Savi, D.C.; Adamoski, D.; Kava, V.M.; Hungria, M.; Galli-Terasawa, L.V. Diversity of maize (Zea mays L.) rhizobacteria with potential to promote plant growth. Braz. J. Microbiol. 2021, 52, 1807–1823. [Google Scholar] [CrossRef]
- Liu, J.; Tang, Y.; Bao, J.; Wang, H.; Peng, F.; Tan, P.; Chu, G.; Liu, S. A stronger rhizosphere impact on the fungal communities compared to the bacterial communities in pecan plantations. Front. Microbiol. 2022, 13, 899801. [Google Scholar] [CrossRef]
- Guerrero-Galán, C.; Calvo-Polanco, M.; Zimmermann, S.D. Ectomycorrhizal symbiosis helps plants to challenge salt stress conditions. Mycorrhiza 2019, 29, 291–301. [Google Scholar] [CrossRef]
- Egidi, E.; Delgado-Baquerizo, M.; Plett, J.M.; Wang, J.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 2019, 10, 2369. [Google Scholar] [CrossRef]
- Ge, Z.-W.; Brenneman, T.; Bonito, G.; Smith, M.E. Soil pH and mineral nutrients strongly influence truffles and other ectomycorrhizal fungi associated with commercial pecans (Carya illinoinensis). Plant Soil 2017, 418, 493–505. [Google Scholar] [CrossRef]
- Uroz, S.; Oger, P.; Tisserand, E.; Cébron, A.; Turpault, M.P.; Buée, M.; De Boer, W.; Leveau, J.H.J.; Frey-Klett, P. Specific impacts of beech and Norway spruce on the structure and diversity of the rhizosphere and soil microbial communities. Sci. Rep. 2016, 6, 27756. [Google Scholar] [CrossRef]
- Jiang, S.; Xing, Y.; Liu, G.; Hu, C.; Wang, X.; Yan, G.; Wang, Q. Changes in soil bacterial and fungal community composition and functional groups during the succession of boreal forests. Soil Biol. Biochem. 2021, 161, 108393. [Google Scholar] [CrossRef]
- Hartmann, M.; Niklaus, P.A.; Zimmermann, S.; Schmutz, S.; Kremer, J.; Abarenkov, K.; Lüscher, P.; Widmer, F.; Frey, B. Resistance and resilience of the forest soil microbiome to logging-associated compaction. ISME J. 2014, 8, 226–244. [Google Scholar] [CrossRef]
- Liu, J.; Tang, Y.; Bao, J.; Wang, H.; Chen, M.; Peng, F.; Tan, P. Integrated microbiome and metabolomics analysis reveal a closer relationship between soil metabolites and bacterial community than fungal community in pecan plantations. Land Degrad. Dev. 2023, 34, 2812–2824. [Google Scholar] [CrossRef]
- Frey, B.; Carnol, M.; Dharmarajah, A.; Brunner, I.; Schleppi, P. Only minor changes in the soil microbiome of a sub-alpine forest after 20 years of moderately increased nitrogen loads. Front. Glob. Chang. 2020, 3, 77. [Google Scholar] [CrossRef]
- Tahovská, K.; Choma, M.; Kaštovská, E.; Oulehle, F.; Bárta, J.; Šantrůčková, H.; Moldan, F. Positive response of soil microbes to long-term nitrogen input in spruce forest: Results from Gårdsjön whole-catchment N-addition experiment. Soil Biol. Biochem. 2020, 143, 107732. [Google Scholar] [CrossRef]
- Zou, M.; Yu, K.; Liu, H.; Sheng, Q.; Zhang, Y. Effects of Bacillus subtilis on rose growth promotion and rhizosphere microbial community changes under saline–alkaline stress. Agronomy 2024, 14, 730. [Google Scholar] [CrossRef]
- Wang, W.; Sun, X.; Huang, W.; Men, X.; Yi, S.; Zheng, F.; Zhang, Z.; Wang, Z. Soil P solubilization and plant growth promotion by a saline–alkali-tolerant P-solubilizing bacterium, Bacillus sp. DYS211. J. Plant Eco. 2023, 16, rtad028. [Google Scholar] [CrossRef]
- Sousa, N.R.; Franco, A.R.; Ramos, M.A.; Oliveira, R.S.; Castro, P.M. The response of Betula pubescens to inoculation with an ectomycorrhizal fungus and a plant growth promoting bacterium is substrate-dependent. Ecol. Eng. 2015, 81, 439–443. [Google Scholar] [CrossRef]
- Aggangan, N.S.; Dell, B.; Malajczuk, N. Effects of soil pH on the ectomycorrhizal response of Eucalyptus urophylla seedlings. New Phytol. 1996, 134, 539–546. [Google Scholar] [CrossRef]
- Thiem, D.; Goebel, M.; Gołębiewski, M.; Baum, C.; Koczorski, P.; Szymańska, S.; Hrynkiewicz, K. Endophytic microbiota and ectomycorrhizal structure of Alnus glutinosa Gaertn. at saline and nonsaline forest sites. Sci. Rep. 2023, 13, 22831. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Ma, L.; Jiao, J.; Fahim, A.M.; Wu, J.; Tao, X.; Lian, Y.; Li, R.; Li, Y.; Yang, G. Impact of straw incorporation on the physicochemical profile and fungal ecology of saline–alkaline soil. Microorganisms 2024, 12, 277. [Google Scholar] [CrossRef] [PubMed]
- Ozimek, E.; Hanaka, A. Mortierella species as the plant growth-promoting fungi present in the agricultural soils. Agriculture 2020, 11, 7. [Google Scholar] [CrossRef]
- Li, Y.; Shen, C.; Wang, Y.; Xu, L.; Zhao, Y.; Yi, S.; Zuo, W.; Yao, R.; Zhang, X.; Gu, C. Alleviated environmental constraints and restructured fungal microbiome facilitate aggregate formation and stabilization in coastal mudflat saline soil amended by sewage sludge. Land Degrad. Dev. 2023, 34, 3064–3075. [Google Scholar] [CrossRef]
- Galardis, M.M.B.; Sánchez, R.C.L.; Fall, F.; Eichler-Löbermann, B.; Pruneau, L.; Bâ, A.M. Growth and physiological responses of ectomycorrhizal Coccoloba uvifera (L.) L. seedlings to salt stress. J. Arid Environ. 2022, 196, 104650. [Google Scholar] [CrossRef]
- Sánchez-Ledesma, J.A.; Arreola-Ávila, J.G.; Ávila-Rodríguez, V.; García-González, F.; Carrasco-Hernández, V.; Borja de laRosa, A. Photosynthetic rate and biomass production by inoculation of Scleroderma sp. with different concentrations of NaCl in pecan tree. Rev. Mex. Cienc. Agríc. 2022, 13, 1209–1220. [Google Scholar]
- Wang, M.; Zheng, Q.; Shen, Q.; Guo, S. The critical role of potassium in plant stress response. Int. J. Mol. Sci. 2013, 14, 7370–7390. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, J.; Li, T.; Shao, P.; Ma, J.; Dong, K. Effects of nitrogen and phosphorus imbalance input on rhizosphere and bulk soil bacterial community of Suaeda salsa in the Yellow River Delta. Front. Mar. Sci. 2023, 10, 1131713. [Google Scholar] [CrossRef]
- Anthony, M.A.; Tedersoo, L.; De Vos, B.; Croisé, L.; Meesenburg, H.; Wagner, M.; Andreae, H.; Jacob, F.; Lech, P.; Kowalska, A.; et al. Fungal community composition predicts forest carbon storage at a continental scale. Nat. Commun. 2024, 15, 2385. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, J.; Chen, R.; Zhong, L.; Lin, X.; Feng, Y. Microbial community composition and activity in saline soils of coastal agro–ecosystems. Microorganisms 2022, 10, 835. [Google Scholar] [CrossRef]
- Yan, Y.; Li, B.; Huang, Z.; Zhang, H.; Wu, X.; Farooq, T.H.; Wu, P.; Li, M.; Ma, X. Characteristics and driving factors of rhizosphere bacterial communities of Chinese fir provenances. Forests 2021, 12, 1362. [Google Scholar] [CrossRef]
- Deng, J.; Yin, Y.; Zhu, W.; Zhou, Y. Variations in soil bacterial community diversity and structures among different revegetation types in the Baishilazi Nature Reserve. Front. Microbiol. 2018, 9, 2874. [Google Scholar] [CrossRef]
- Zi, H.; Jiang, Y.; Cheng, X.; Li, W.; Huang, X. Change of rhizospheric bacterial community of the ancient wild tea along elevational gradients in Ailao mountain, China. Sci. Rep. 2020, 10, 9203. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, F.; Sun, Y.; Yang, Z.; Li, R.; Bai, G.; Lin, W.; Chen, H. Appropriately reduced nitrogen and increased phosphorus in ratooning rice increased the yield and reduced the greenhouse gas emissions in southeast china. Plants 2024, 13, 438. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Physicochemical Properties | DF | GM |
|---|---|---|
| TN (g/kg) | 0.42 ± 0.02 b | 0.65 ± 0.02 a |
| TP (g/kg) | 0.66 ± 0.03 a | 0.48 ± 0.01 b |
| TK (g/kg) | 13.58 ± 0.45 a | 9.49 ± 0.56 b |
| AN (mg/kg) | 36.27 ± 1.02 b | 69.74 ± 2.35 a |
| AP (mg/kg) | 38.83 ± 0.15 b | 62.06 ± 4.95 a |
| AK (mg/kg) | 93.58 ± 0.05 b | 117.26 ± 5.84 a |
| SOM (g/kg) | 8.44 ± 0.33 b | 12.94 ± 0.14 a |
| TSS (g/kg) | 0.86 ± 0.24 a | 0.37 ± 0.03 b |
| pH | 8.04 ± 0.02 a | 5.60 ± 0.41 b |
| EC (μs/cm) | 220.00 ± 3.61 a | 121.87 ± 15.55 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Chen, T.; Chen, Y.; Li, S.; Wang, W.; Zhao, Y.; Zhu, C. A Comparative Analysis of Bacterial and Fungal Communities in Coastal and Inland Pecan Plantations. Microorganisms 2024, 12, 1313. https://doi.org/10.3390/microorganisms12071313
Zhang S, Chen T, Chen Y, Li S, Wang W, Zhao Y, Zhu C. A Comparative Analysis of Bacterial and Fungal Communities in Coastal and Inland Pecan Plantations. Microorganisms. 2024; 12(7):1313. https://doi.org/10.3390/microorganisms12071313
Chicago/Turabian StyleZhang, Shijie, Ting Chen, Yu Chen, Shucheng Li, Wu Wang, Yuqiang Zhao, and Cancan Zhu. 2024. "A Comparative Analysis of Bacterial and Fungal Communities in Coastal and Inland Pecan Plantations" Microorganisms 12, no. 7: 1313. https://doi.org/10.3390/microorganisms12071313
APA StyleZhang, S., Chen, T., Chen, Y., Li, S., Wang, W., Zhao, Y., & Zhu, C. (2024). A Comparative Analysis of Bacterial and Fungal Communities in Coastal and Inland Pecan Plantations. Microorganisms, 12(7), 1313. https://doi.org/10.3390/microorganisms12071313