
A CRISPR-Cas9-Mediated Large-Fragment Assembly Method for Cloning Genomes and Biosynthetic Gene Cluster


Abstract
:1. Introduction
2. Materials and Method
2.1. Synthesis and Purification of sgRNA
2.2. Expression and Purification of Cas9
2.3. Preparation of Genomic DNA
2.4. In Vitro Cas9 Endonuclease Cleavage for Genomic DNA
2.5. DNA Purification after In Vitro Cas9 Digestion
2.6. Preparation of Vector with the Homologous Arm
2.7. Gibson Assembly
2.8. Dialysis
2.9. Electroporation
3. Results
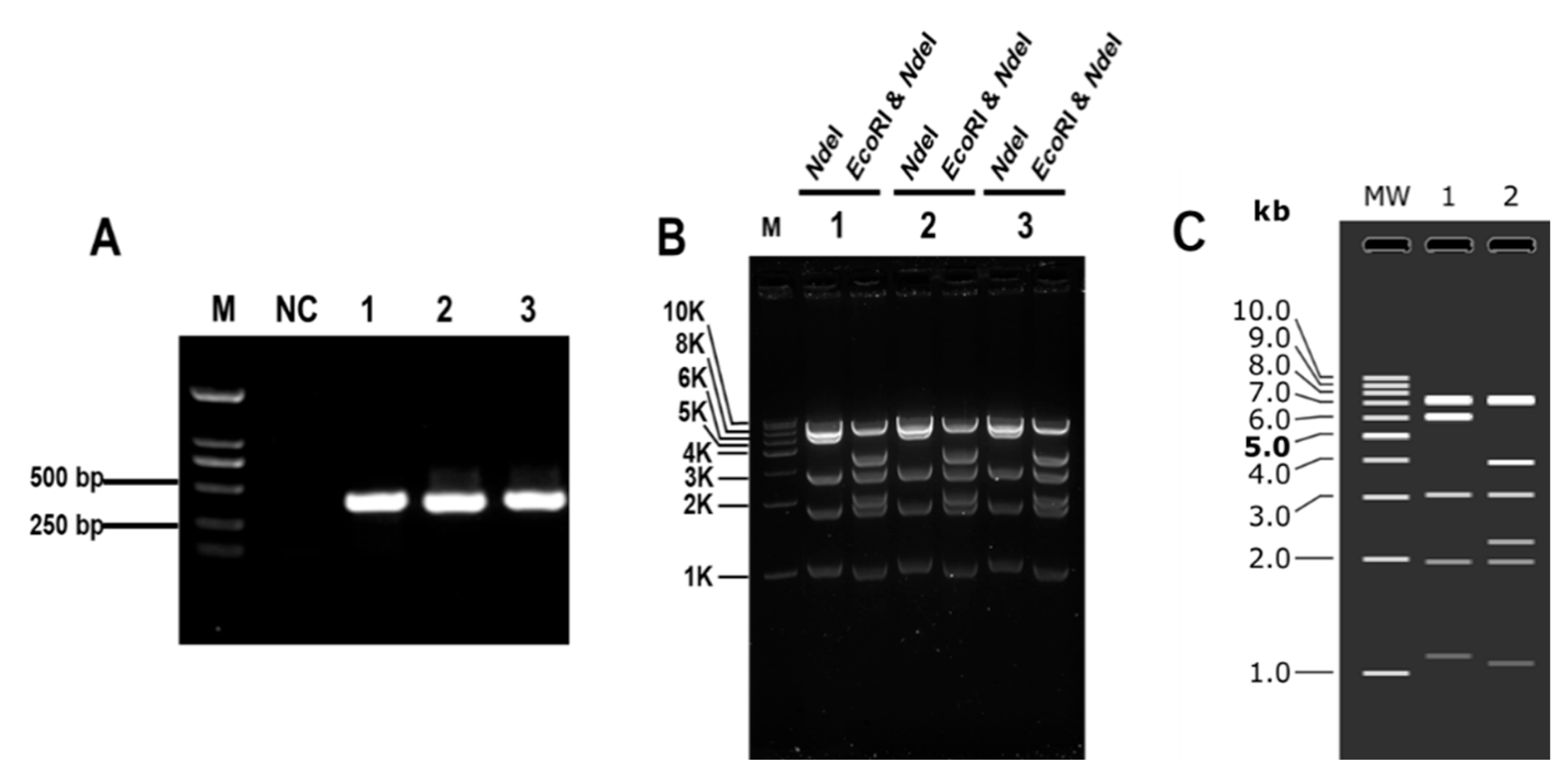
3.1. Establishment of CRISPR/Cas9 System for Cloning Large DNA Fragments In Vitro
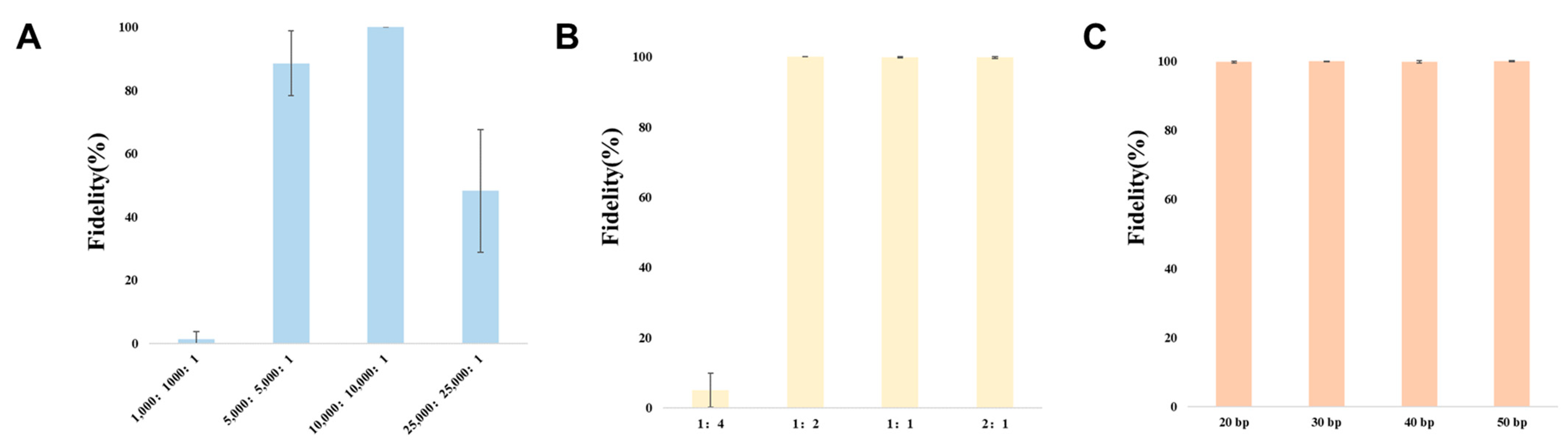
3.2. Optimization of CRISPR/Cas9 System for Cloning Large DNA Fragments In Vitro
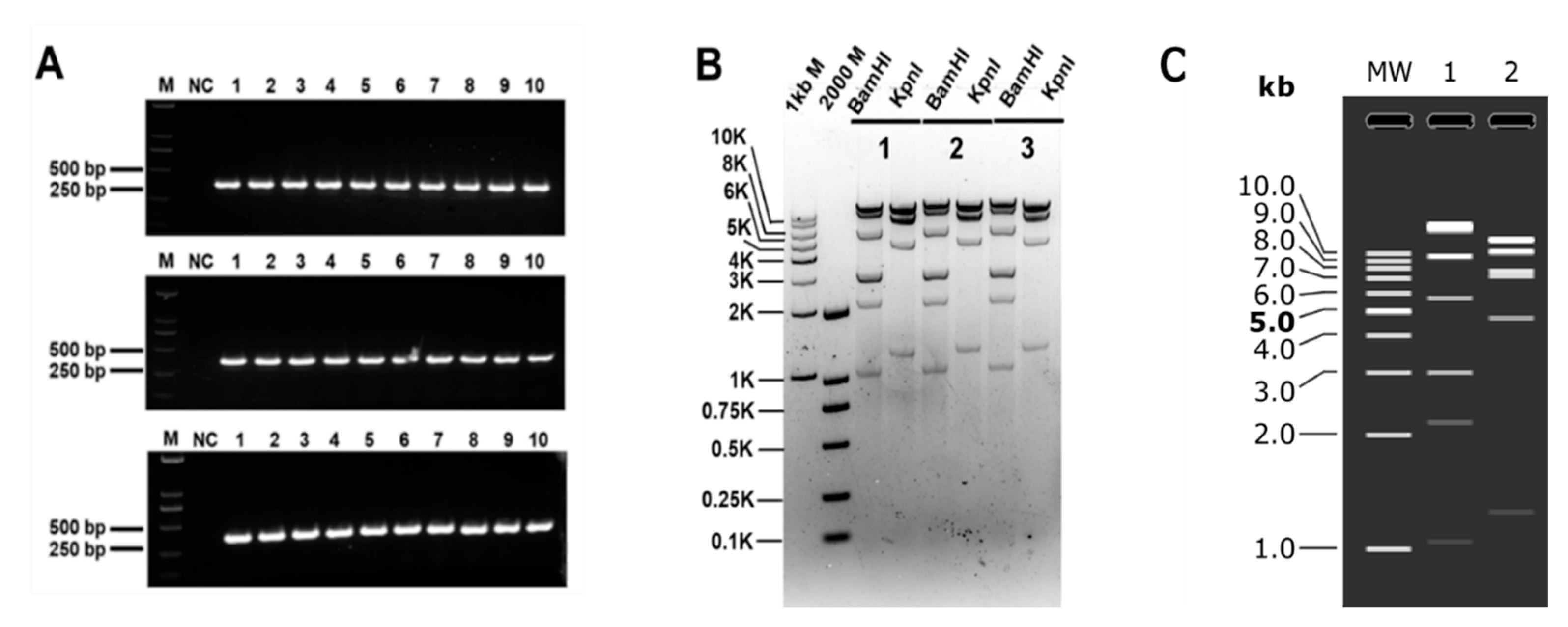
3.3. Clones of Large DNA Fragments of Different Lengths
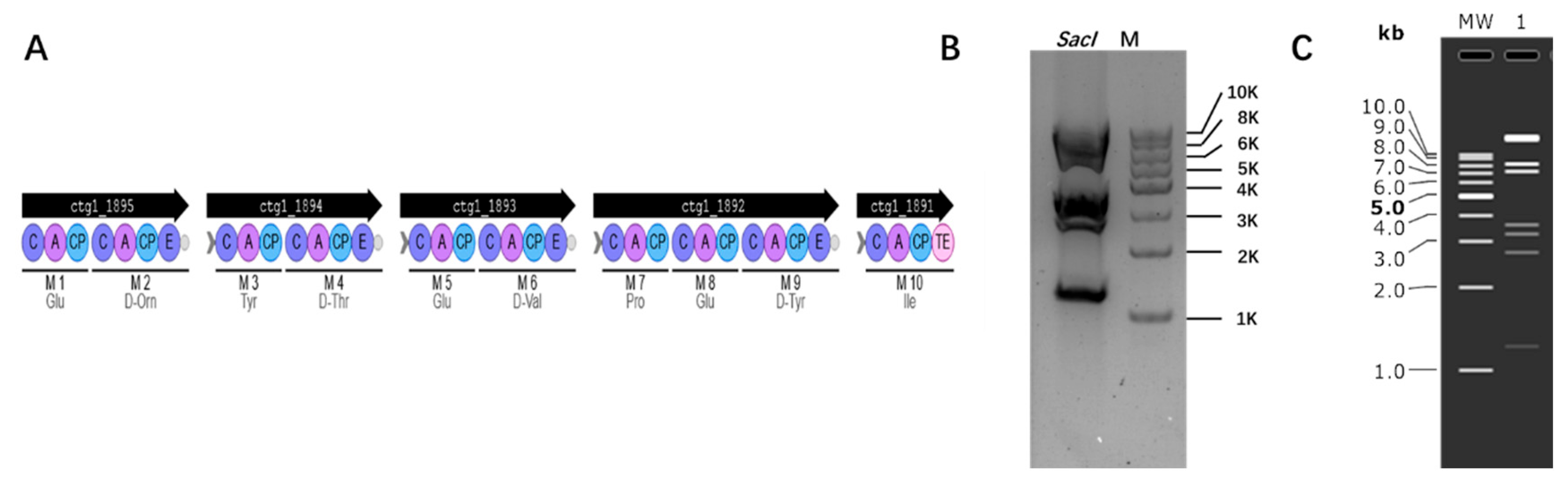
3.4. Cloning Biosynthetic Gene Cluster
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ellis, T.; Adie, T.; Baldwin, G.S. DNA assembly for synthetic biology: From parts to pathways and beyond. Integr. Biol. Quant. Biosci. Nano Macro 2011, 3, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, R.; Xue, X.; Qin, Z. CasHRA (Cas9-facilitated Homologous Recombination Assembly) method of constructing megabase-sized DNA. Nucleic Acids Res. 2016, 44, e124. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.X.; Liu, D.; Li, B.Z.; Zhao, M.; Zeng, B.X.; Wu, Y.; Shen, Y.; Lin, T.; Yang, P.; Dai, J.; et al. Design and chemical synthesis of eukaryotic chromosomes. Chem. Soc. Rev. 2017, 46, 7191–7207. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, D.; Shen, Y. Discovery of novel bioactive natural products driven by genome mining. Drug Discov. Ther. 2018, 12, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, E.; Nakagawa, A.; Tomabechi, Y.; Ikushiro, S.; Sakaki, T.; Katayama, T.; Yamamoto, K.; Kumagai, H.; Sato, F.; Minami, H. Microbial production of novel sulphated alkaloids for drug discovery. Sci. Rep. 2018, 8, 7980. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Kim, E.S. Recent advances in heterologous expression of natural product biosynthetic gene clusters in Streptomyces hosts. Curr. Opin. Biotechnol. 2021, 69, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Fang, Y.; Ding, M.; Zhang, Y.; Jia, K.; Li, Z.; Collemare, J.; Liu, W. Developing fungal heterologous expression platforms to explore and improve the production of natural products from fungal biodiversity. Biotechnol. Adv. 2022, 54, 107866. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Feng, J.; Zhang, P.; Fan, J.; Yin, W.B. A CRISPR/Cas9 Cleavage System for Capturing Fungal Secondary Metabolite Gene Clusters. J. Microbiol. Biotechnol. 2021, 31, 8–15. [Google Scholar] [CrossRef]
- Bian, X.; Huang, F.; Stewart, F.A.; Xia, L.; Zhang, Y.; Müller, R. Direct cloning, genetic engineering, and heterologous expression of the syringolin biosynthetic gene cluster in E. coli through Red/ET recombineering. Chembiochem A Eur. J. Chem. Biol. 2012, 13, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Tang, X.; Moore, B.S. Genetic platforms for heterologous expression of microbial natural products. Nat. Prod. Rep. 2019, 36, 1313–1332. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, A.; Liu, J.; Huang, W.; Wang, J.; Cai, Z.; Zhao, G. iCatch: A new strategy for capturing large DNA fragments using homing endonucleases. Acta Biochim. Biophys. Sin. 2019, 51, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, W.; Wen, S.; Li, Z.; Yang, A.; Deng, Z.; Sun, Y. In Vitro CRISPR/Cas9 System for Efficient Targeted DNA Editing. mBio 2015, 6, e01714–e01715. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Chen, L.; Zhao, C.; Wu, J.; Yan, D.; Deng, Z.; Sun, Y. In Vitro Packaging Mediated One-Step Targeted Cloning of Natural Product Pathway. ACS Synth. Biol. 2019, 8, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Nielsen, J.; Liu, Z. Bioprospecting Through Cloning of Whole Natural Product Biosynthetic Gene Clusters. Front. Bioeng. Biotechnol. 2020, 8, 526. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Kim, J.H.; Larionov, V. Highly Selective, CRISPR/Cas9-Mediated Isolation of Genes and Genomic Loci from Complex Genomes by TAR Cloning in Yeast. Curr. Protoc. 2021, 1, e207. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Larionov, V.; Kouprina, N. Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res. 2015, 43, e55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Jia, R.; Yin, J.; Li, A.; Xia, L.; Yin, Y.; Müller, R.; Fu, J.; Stewart, A.F.; et al. ExoCET: Exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Res. 2018, 46, e28. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Jia, R.; Hou, Y.; Yin, J.; Bian, X.; Li, A.; Müller, R.; Stewart, A.F.; Fu, J.; et al. RecET direct cloning and Redαβ recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nat. Protoc. 2016, 11, 1175–1190. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bian, X.; Hu, S.; Wang, H.; Huang, F.; Seibert, P.M.; Plaza, A.; Xia, L.; Müller, R.; Stewart, A.F.; et al. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 2012, 30, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhao, X.; Gabrieli, T.; Lou, C.; Ebenstein, Y.; Zhu, T.F. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun. 2015, 6, 8101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, T.F. Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat. Protoc. 2016, 11, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Liu, L.; Xu, F.; Zeng, X.; Wang, R.; Yang, J.; Wang, W.; Karthik, L.; Liu, J.; Yang, Z.; et al. Activating cryptic biosynthetic gene cluster through a CRISPR-Cas12a-mediated direct cloning approach. Nucleic Acids Res. 2022, 50, 3581–3592. [Google Scholar] [CrossRef] [PubMed]
- Clos, J.; Zander-Dinse, D. Cosmid Library Construction and Functional Cloning. Methods Mol. Biol. 2019, 1971, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Enghiad, B.; Huang, C.; Guo, F.; Jiang, G.; Wang, B.; Tabatabaei, S.K.; Martin, T.A.; Zhao, H. Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination. Nat. Commun. 2021, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Liu, Z.; Zhang, X.; Zhang, G.; Xie, Y.; Ding, X.; Mo, X.; Stewart, A.F.; Fu, J.; Zhang, Y.; et al. “Cre/loxP plus BAC”: A strategy for direct cloning of large DNA fragment and its applications in Photorhabdus luminescens and Agrobacterium tumefaciens. Sci. Rep. 2016, 6, 29087. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang, L.; Tian, Y.; Liu, H.; Tan, H.; Niu, G. Genome engineering and direct cloning of antibiotic gene clusters via phage ϕBT1 integrase-mediated site-specific recombination in Streptomyces. Sci. Rep. 2015, 5, 8740. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.Z.; Zhou, J.T.; Li, B.Z.; Yuan, Y.J. The TelN/tos-assisted precise targeting of chromosome segments (TAPE). J. Adv. Res. 2022, 41, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lim, H.; Jang, H.; Hwang, B.; Lee, J.H.; Cho, J.; Lee, J.H.; Bang, D. CRISPR-Cap: Multiplexed double-stranded DNA enrichment based on the CRISPR system. Nucleic Acids Res. 2019, 47, e1. [Google Scholar] [CrossRef] [PubMed]
- Vahidinasab, M.; Adiek, I.; Hosseini, B.; Akintayo, S.O.; Abrishamchi, B.; Pfannstiel, J.; Henkel, M.; Lilge, L.; Voegele, R.T.; Hausmann, R. Characterization of Bacillus velezensis UTB96, Demonstrating Improved Lipopeptide Production Compared to the Strain B. velezensis FZB42. Microorganisms 2022, 10, 2225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Scheuring, C.F.; Wu, C.C.; Dong, J.J.; Zhang, H.B. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat. Protoc. 2012, 7, 467–478. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | Disadvantages | DNA Fragments | Fidelity | Cycle | |
|---|---|---|---|---|---|
| LLHR [21] | Technically easier; suitable for cloning small- and mid-BGCs; simple to use for recombination with short homologous arms. | False positives; difficult to clone large-size BGCs. | <~52 kb | <~50% | ~3 d |
| ExoCET [19] | Technically easier; simple to use for recombination with short homologous arms. | Low efficiency for cloning large-size BGCs. | <~106 kb | 4~100% | ~3 d |
| TAR-CRISPR [18] | Cas9-facilitated high-efficiency cloning; suitable for cloning large genomic regions. | Technically challenging to use yeast spheroplasts for highly efficient transformation; some false positives; requires careful preparation and/or manipulation of gDNA. | - | <35% | ~7 d |
| CATCH [22] | Suitable for cloning large genomic regions. | Requires careful preparation of the genomic DNA in gel. | <~150 kb | 2~90% | ~4 d |
| CAT-FISHING [24] | Suitable for cloning large genomic regions with high GC. | Low efficiency. | <~145 kb | 8~55% | 3~4 d |
| This method | Technically easier; short cycle; high fidelity; can clone large fragments from different sources. | <~80 kb | 46–100% | ~2.5 d |
| Group | Efficiency | Fidelity | Average Fidelity | |
|---|---|---|---|---|
| 15 kb | 1 | 448/1 | 99.8% | 99.9% |
| 2 | 628/0 | 100% | ||
| 3 | 366/0 | 100% | ||
| 30 kb | 1 | 124/3 | 97.6% | 99.0% |
| 2 | 79/0 | 100% | ||
| 3 | 179/1 | 99.4% | ||
| 50 kb | 1 | 46/0 | 100% | 97.7% |
| 2 | 37/1 | 97.4% | ||
| 3 | 67/3 | 95.7% | ||
| 60 kb | 1 | 12/14 | 46.1% | 62.4% |
| 2 | 18/8 | 69.2% | ||
| 3 | 23/9 | 71.9% | ||
| 77 kb | 1 | 9/6 | 60% | 46.0% |
| 2 | 15/14 | 44.8% | ||
| 3 | 6/12 | 33.3% | ||
| 100 kb | 1 | 0/0 | 0 | N/A |
| 2 | 0/12 | 0 | ||
| 3 | 0/6 | 0 |
| Group | Efficiency | Fidelity | Average Fidelity |
|---|---|---|---|
| 1 | 27/30 | 90% | 93% |
| 2 | 39/42 | 92.8% | |
| 3 | 19/20 | 95% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Cai, G.; Li, H.; Lin, Z.; Shi, S.; Jin, J.; Liu, Z. A CRISPR-Cas9-Mediated Large-Fragment Assembly Method for Cloning Genomes and Biosynthetic Gene Cluster. Microorganisms 2024, 12, 1462. https://doi.org/10.3390/microorganisms12071462
Guo Y, Cai G, Li H, Lin Z, Shi S, Jin J, Liu Z. A CRISPR-Cas9-Mediated Large-Fragment Assembly Method for Cloning Genomes and Biosynthetic Gene Cluster. Microorganisms. 2024; 12(7):1462. https://doi.org/10.3390/microorganisms12071462
Chicago/Turabian StyleGuo, Yujing, Guang Cai, Huiying Li, Zhenquan Lin, Shuobo Shi, Jin Jin, and Zihe Liu. 2024. "A CRISPR-Cas9-Mediated Large-Fragment Assembly Method for Cloning Genomes and Biosynthetic Gene Cluster" Microorganisms 12, no. 7: 1462. https://doi.org/10.3390/microorganisms12071462
APA StyleGuo, Y., Cai, G., Li, H., Lin, Z., Shi, S., Jin, J., & Liu, Z. (2024). A CRISPR-Cas9-Mediated Large-Fragment Assembly Method for Cloning Genomes and Biosynthetic Gene Cluster. Microorganisms, 12(7), 1462. https://doi.org/10.3390/microorganisms12071462