
Microbial Metagenomics Revealed the Diversity and Distribution Characteristics of Groundwater Microorganisms in the Middle and Lower Reaches of the Yangtze River Basin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
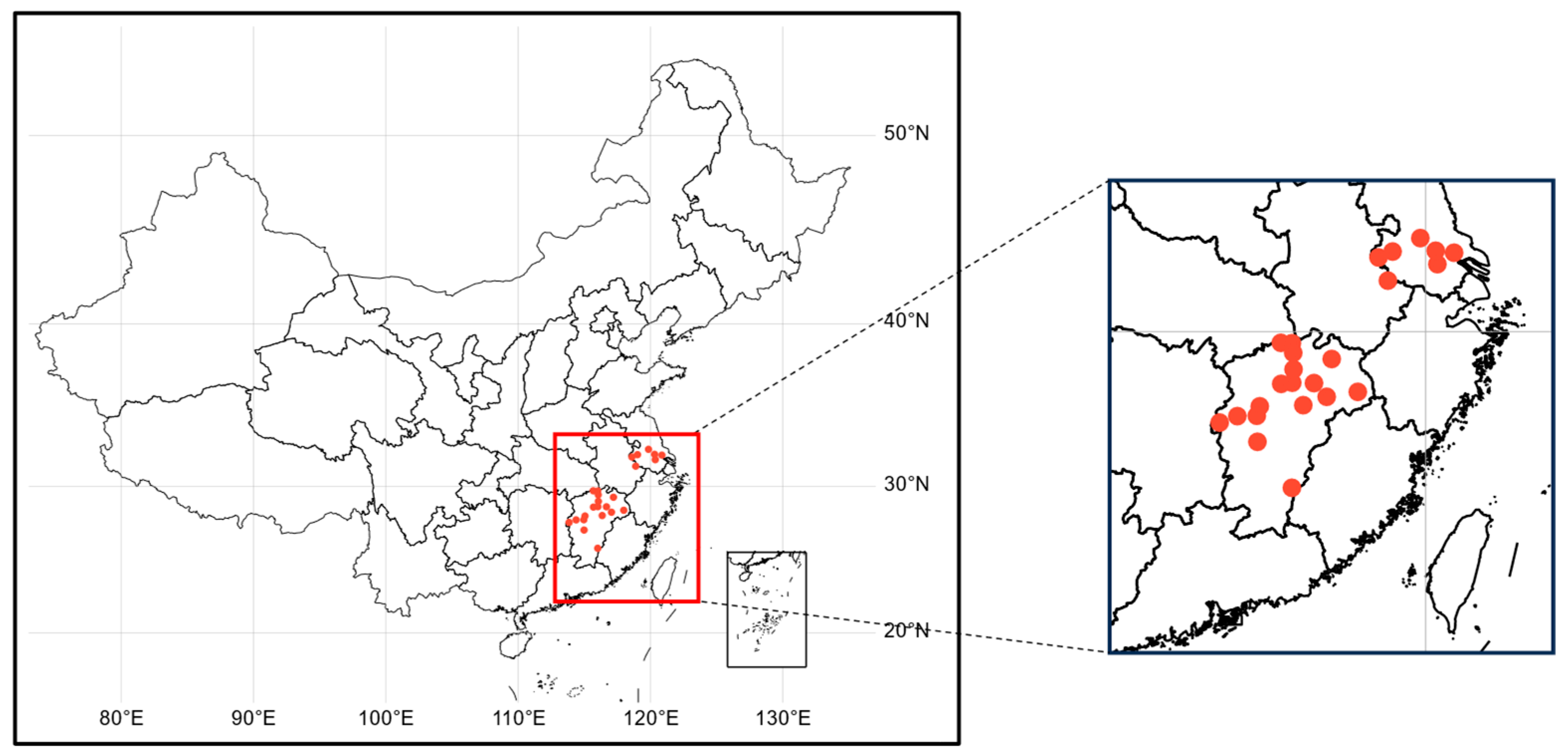
2.1. Sample Collection
2.2. DNA Extraction, Metagenomic Sequencing, and Quality Analysis
2.3. Bioinformatic Analysis
2.4. Statistical Analysis
3. Results
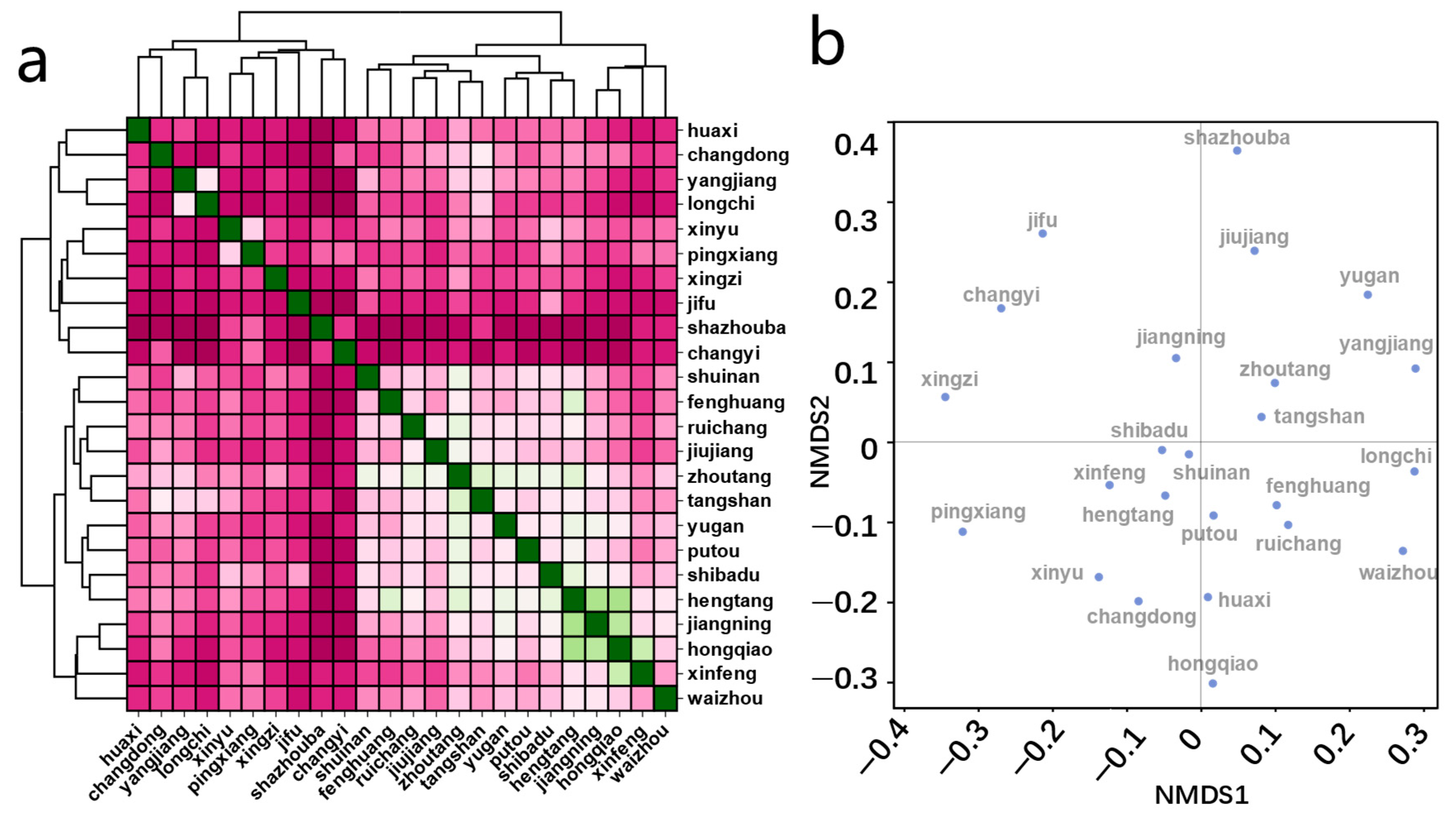
3.1. Distribution and Composition of Groundwater Microbial Communities
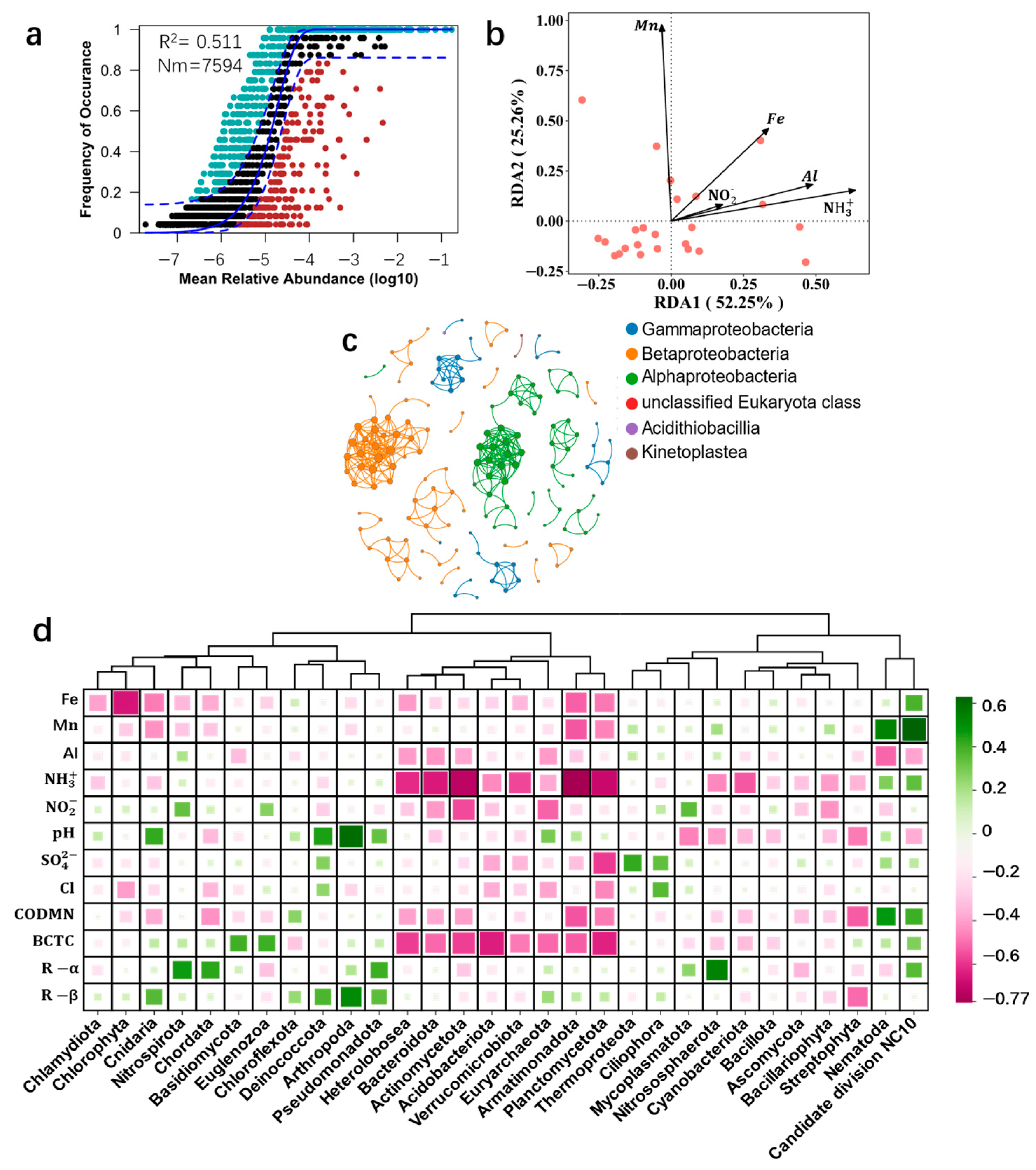
3.2. Microbial Community Response to Environmental Factors
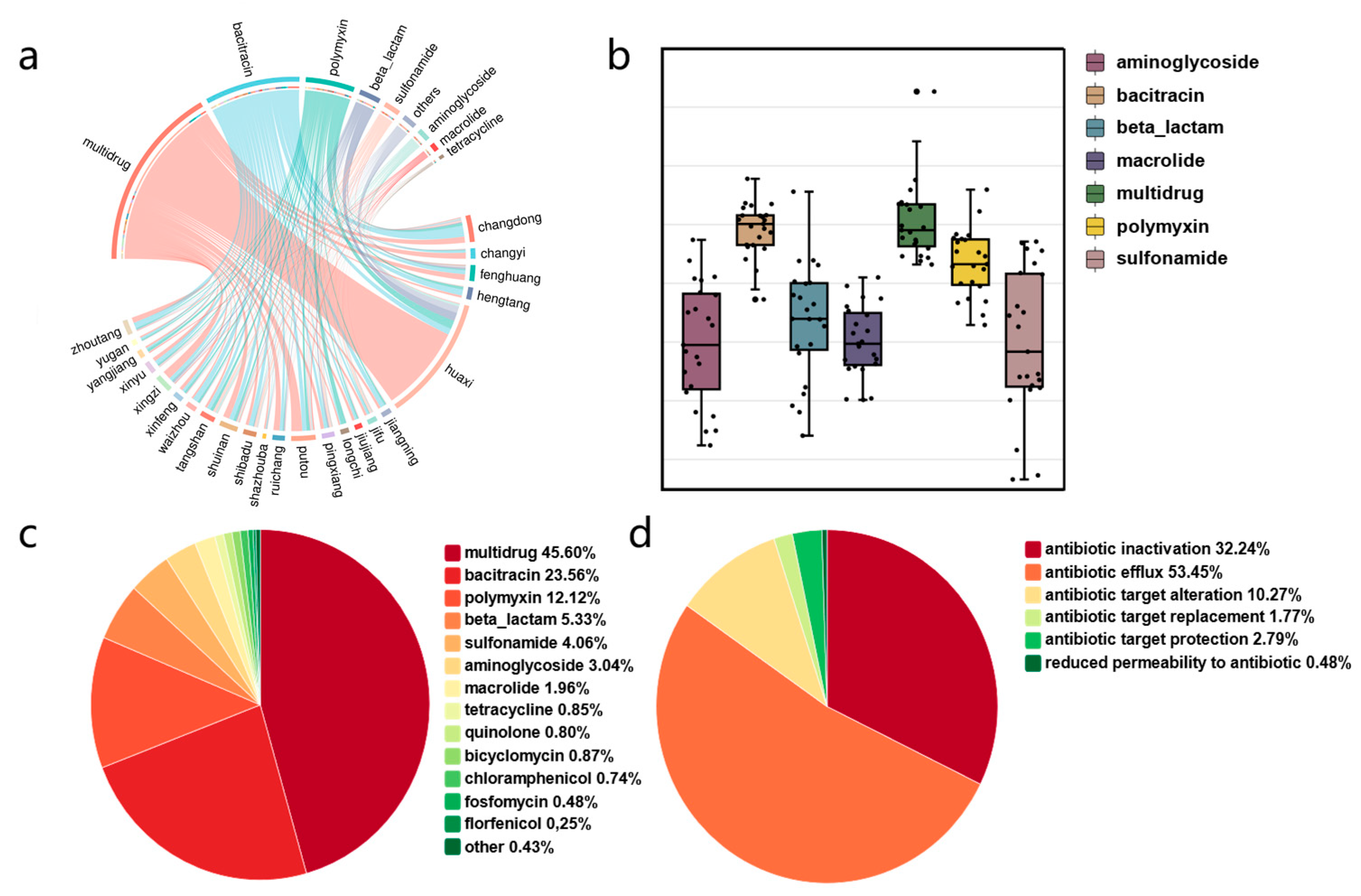
3.3. Mechanisms of Antibiotic Resistance
3.4. Mobility Potential of Antibiotic Resistance Genes in Groundwater
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Azam, F. Microbial control of oceanic carbon flux: The plot thickens. Science 1998, 280, 694–696. [Google Scholar] [CrossRef]
- Gleick, P.H. Water in Crisis: A Guide to the World’s Fresh Water Resources; Oxford University Press: New York, NY, USA, 1993. [Google Scholar]
- Griebler, C.; Lueders, T. Microbial biodiversity in groundwater ecosystems. Freshwater Biol. 2009, 54, 649–677. [Google Scholar] [CrossRef]
- Hemme, C.L.; Tu, Q.; Shi, Z.; Qin, Y.; Gao, W.; Deng, Y.; Nostrand, J.D.; Wu, L.; He, Z.; Chain, P.S.; et al. Comparative metagenomics reveals impact of contaminants on groundwater microbiomes. Front. Microbiol. 2015, 6, 1205. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Ju, F.; Zhang, T. Experimental Design and Bioinformatics Analysis for the Application of Metagenomics in Environmental Sciences and Biotechnology. Environ. Sci. Technol. 2015, 49, 12628–12640. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Qin, Y.; Chen, T.; Lu, M.P.; Qian, X.B.; Guo, X.X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Tomofuji, Y.; Kishikawa, T.; Maeda, Y.; Ogawa, K.; Otake-Kasamoto, Y.; Kawabata, S.; Nii, T.; Okuno, T.; Oguro-Igashira, E.; Kinoshita, M.; et al. Prokaryotic and viral genomes recovered from 787 Japanese gut metagenomes revealed microbial features linked to diets, populations, and diseases. Cell Genom. 2022, 2, 100219. [Google Scholar] [CrossRef]
- Salazar, G.; Paoli, L.; Alberti, A.; Huerta-Cepas, J.; Ruscheweyh, H.J.; Cuenca, M.; Field, C.M.; Coelho, L.P.; Cruaud, C.; Engelen, S.; et al. Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome. Cell 2019, 179, 1068–1683. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kaown, D.; Mayer, B.; Lee, J.Y.; Hyun, Y.; Lee, K.K. Identifying the sources of nitrate contamination of groundwater in an agricultural area (Haean basin, Korea) using isotope and microbial community analyses. Sci. Total Environ. 2015, 533, 566–575. [Google Scholar] [CrossRef]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef]
- Moore, A.; Lenczewski, M.; Leal-Bautista, R.M.; Duvall, M. Groundwater microbial diversity and antibiotic resistance linked to human population density in Yucatan Peninsula, Mexico. Can. J. Microbiol. 2020, 66, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Savio, D.; Stadler, P.; Reischer, G.H.; Demeter, K.; Linke, R.B.; Blaschke, A.P.; Mach, R.L.; Kirschner, A.K.T.; Stadler, H.; Farnleitner, A.H. Spring Water of an Alpine Karst Aquifer Is Dominated by a Taxonomically Stable but Discharge-Responsive Bacterial Community. Front. Microbiol. 2019, 10, 28. [Google Scholar] [CrossRef]
- Alfreider, A.; Vogt, C. Genetic Evidence for Bacterial Chemolithoautotrophy Based on the Reductive Tricarboxylic Acid Cycle in Groundwater Systems. Microbes Environ. 2012, 27, 209–214. [Google Scholar] [CrossRef]
- Wegner, C.E.; Gaspar, M.; Geesink, P.; Herrmann, M.; Marz, M.; Küsel, K. Erratum for Wegner et al., “Biogeochemical Regimes in Shallow Aquifers Reflect the Metabolic Coupling of the Elements Nitrogen, Sulfur, and Carbon”. Appl. Environ. Microb. 2019, 85, e02346-18. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.C.; Fredrickson, J.K.; Wilkins, M.J.; Konopka, A.E.; Nelson, W.C.; Arntzen, E.V.; Chrisler, W.B.; Chu, R.K.; Danczak, R.E.; Fansler, S.J.; et al. Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat. Commun. 2016, 7, 11237. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.Y.; Zhang, L.P.; Li, F.R.; Huang, S.J.; Yang, J.; Ma, C.; Zhang, D.X.; Yu, C.P.; Hu, A.Y. Urban ponds as hotspots of antibiotic resistome in the urban environment. J. Hazard. Mater. 2021, 403, 124008. [Google Scholar] [CrossRef] [PubMed]
- Fresia, P.; Antelo, V.; Salazar, C.; Giménez, M.; D’Alessandro, B.; Afshinnekoo, E.; Mason, C.; Gonnet, G.H.; Iraola, G. Urban metagenomics uncover antibiotic resistance reservoirs in coastal beach and sewage waters. Microbiome 2019, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Michalsen, M.M.; King, A.S.; Istok, J.D.; Crocker, F.H.; Fuller, M.E.; Kucharzyk, K.H.; Gander, M.J. Spatially-distinct redox conditions and degradation rates following field-scale bioaugmentation for RDX-contaminated groundwater remediation. J. Hazard. Mater. 2020, 387, 121529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.G.; Qiu, R.; Lu, L.; Chen, X.; He, C.; Lu, J.P.; Ren, Z.J. Autotrophic Vanadium(V) Bioreduction in Groundwater by Elemental Sulfur and Zerovalent Iron. Environ. Sci. Technol. 2018, 52, 7434–7442. [Google Scholar] [CrossRef]
- Hassell, J.M.; Ward, M.J.; Muloi, D.; Bettridge, J.M.; Phan, H.; Robinson, T.P.; Ogendo, A.; Imboma, T.; Kiiru, J.; Kariuki, S.; et al. Deterministic processes structure bacterial genetic communities across an urban landscape. Nat. Commun. 2019, 10, 2643. [Google Scholar] [CrossRef]
- Hou, L.; Mulla, S.I.; Nino-Garcia, J.P.; Ning, D.; Rashid, A.; Hu, A.; Yu, C.P. Deterministic and stochastic processes driving the shift in the prokaryotic community composition in wastewater treatment plants of a coastal Chinese city. Appl. Microbiol. Biotechnol. 2019, 103, 9155–9168. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Li, S.; Zhang, L.; Wang, H.; Yang, J.; Luo, Z.; Rashid, A.; Chen, S.; Huang, W.; Yu, C.P. Prokaryotic footprints in urban water ecosystems: A case study of urban landscape ponds in a coastal city, China. Environ. Pollut. 2018, 242, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Guo, Y.Y.; Isabwe, A.; Chen, H.H.; Wang, Y.M.; Zhang, Y.P.; Zhu, Z.X.; Yang, J. Urbanization drives riverine bacterial antibiotic resistome more than taxonomic community at watershed scale. Environ. Int. 2020, 137, 105524. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Liu, C.M.; Luo, R.B.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.H.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Jia, B.F.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.Y.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Yin, X.L.; Zheng, X.W.; Li, L.G.; Zhang, A.N.; Jiang, X.T.; Zhang, T. ARGs-OAP v3.0: Antibiotic-Resistance Gene Database Curation and Analysis Pipeline Optimization. Engineering 2023, 27, 234–241. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J. Vegan: R functions for Vegetation Ecologists. 2005. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 5 June 2024).
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Ubbell, S.P. The Unified Neutral Theory of Biodiversity and Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [PubMed]
- Getino, M.; de da Cruz, F. Natural and Artificial Strategies To Control the Conjugative Transmission of Plasmids. Microbiol. Spectr. 2018, 6, mtbp-0015-2016. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Gao, Q.X.; Li, Y.L.; Qi, Z.H.; Yue, Y.F.; Min, M.H.; Peng, S.M.; Shi, Z.H.; Gao, Y. Diverse and abundant antibiotic resistance genes from mariculture sites of China’s coastline. Sci. Total Environ. 2018, 630, 117–125. [Google Scholar] [CrossRef]
- Niu, Z.G.; Zhang, K.; Zhang, Y. Occurrence and distribution of antibiotic resistance genes in the coastal area of the Bohai Bay, China. Mar. Pollut. Bull. 2016, 107, 245–250. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Niu, Z.G.; Zhang, Y.; Zhang, K. Occurrence of intracellular and extracellular antibiotic resistance genes in coastal areas of Bohai Bay (China) and the factors affecting them. Environ. Pollut. 2018, 236, 126–136. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, X.; Jia, D.; Lyu, Y.; Hu, J.; Chen, Q.; Sun, W. Sediments alleviate the inhibition effects of antibiotics on denitrification: Functional gene, microbial community, and antibiotic resistance gene analysis. Sci. Total Environ. 2022, 804, 150092. [Google Scholar] [CrossRef]
- Wang, J.; Fu, B.J.; Qiu, Y.; Chen, L.D. Soil nutrients in relation to land use and landscape position in the semi-arid small catchment on the loess plateau in China. J. Arid. Environ. 2001, 48, 537–550. [Google Scholar] [CrossRef]
- Chen, B.B.; Jiao, S.; Luo, S.W.; Ma, B.B.; Qi, W.; Cao, C.D.; Zhao, Z.G.; Du, G.Z.; Ma, X.J. High soil pH enhances the network interactions among bacterial and archaeal microbiota in alpine grasslands of the Tibetan Plateau. Environ. Microbiol. 2021, 23, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.H.; Tang, Y.; Cai, L.; Yang, Z.H.; Chen, X.P.; Ouyang, Y.R.; Dai, J.J.; Yang, M. Co-Occurrence Relationship and Stochastic Processes Affect Sedimentary Archaeal and Bacterial Community Assembly in Estuarine-Coastal Margins. Microorganisms 2022, 10, 1339. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Liu, N.; Yang, M.L.; Wang, L.J.; Liang, X.; Liu, C.Q. Co-occurrence of planktonic bacteria and archaea affects their biogeographic patterns in China’s coastal wetlands. Environ. Microbiome 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Lin, Y.H.; Liu, T.; Xu, X.M.; Wang, J.W.; Chen, Q.; Sun, W.L.; Dang, C.Y.; Ni, J.R. Planktonic/benthic Bathyarchaeota as a “gatekeeper” enhance archaeal nonrandom co-existence and deterministic assembling in the Yangtze River. Water Res. 2023, 247, 120829. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, A.N.; Wang, J.W.; Liu, S.F.; Jiang, X.T.; Dang, C.Y.; Ma, T.; Liu, S.T.; Chen, Q.; Xie, S.G.; et al. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.M.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. Isme J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef]
- Logares, R.; Lindström, E.S.; Langenheder, S.; Logue, J.B.; Paterson, H.; Laybourn-Parry, J.; Rengefors, K.; Tranvik, L.; Bertilsson, S. Biogeography of bacterial communities exposed to progressive long-term environmental change. Isme J. 2013, 7, 937–948. [Google Scholar] [CrossRef]
- Cheng, M.; Luo, S.; Zhang, P.; Xiong, G.; Chen, K.; Jiang, C.; Yang, F.; Huang, H.; Yang, P.; Liu, G.; et al. A genome and gene catalog of the aquatic microbiomes of the Tibetan Plateau. Nat. Commun. 2024, 15, 1438. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Weng, M.-Y.; Zhong, J.-W.; He, L.; Guo, D.-J.; Luo, D.; Xue, J.-Y. Microbial Metagenomics Revealed the Diversity and Distribution Characteristics of Groundwater Microorganisms in the Middle and Lower Reaches of the Yangtze River Basin. Microorganisms 2024, 12, 1551. https://doi.org/10.3390/microorganisms12081551
Wang Y, Weng M-Y, Zhong J-W, He L, Guo D-J, Luo D, Xue J-Y. Microbial Metagenomics Revealed the Diversity and Distribution Characteristics of Groundwater Microorganisms in the Middle and Lower Reaches of the Yangtze River Basin. Microorganisms. 2024; 12(8):1551. https://doi.org/10.3390/microorganisms12081551
Chicago/Turabian StyleWang, Yue, Ming-Yu Weng, Ji-Wen Zhong, Liang He, De-Jun Guo, Dong Luo, and Jia-Yu Xue. 2024. "Microbial Metagenomics Revealed the Diversity and Distribution Characteristics of Groundwater Microorganisms in the Middle and Lower Reaches of the Yangtze River Basin" Microorganisms 12, no. 8: 1551. https://doi.org/10.3390/microorganisms12081551