
Lysine Phoshoglycerylation Is Widespread in Bacteria and Overlaps with Acylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Culture Conditions
2.2. Sample Preparation for Proteomics
2.3. Mass Spectrometry
2.4. Data Processing and Peptide and Protein Identification
2.5. Re-Analysis of Phosphoproteome Datasets from Other Bacteria
2.6. Additional Methods
3. Results and Discussion
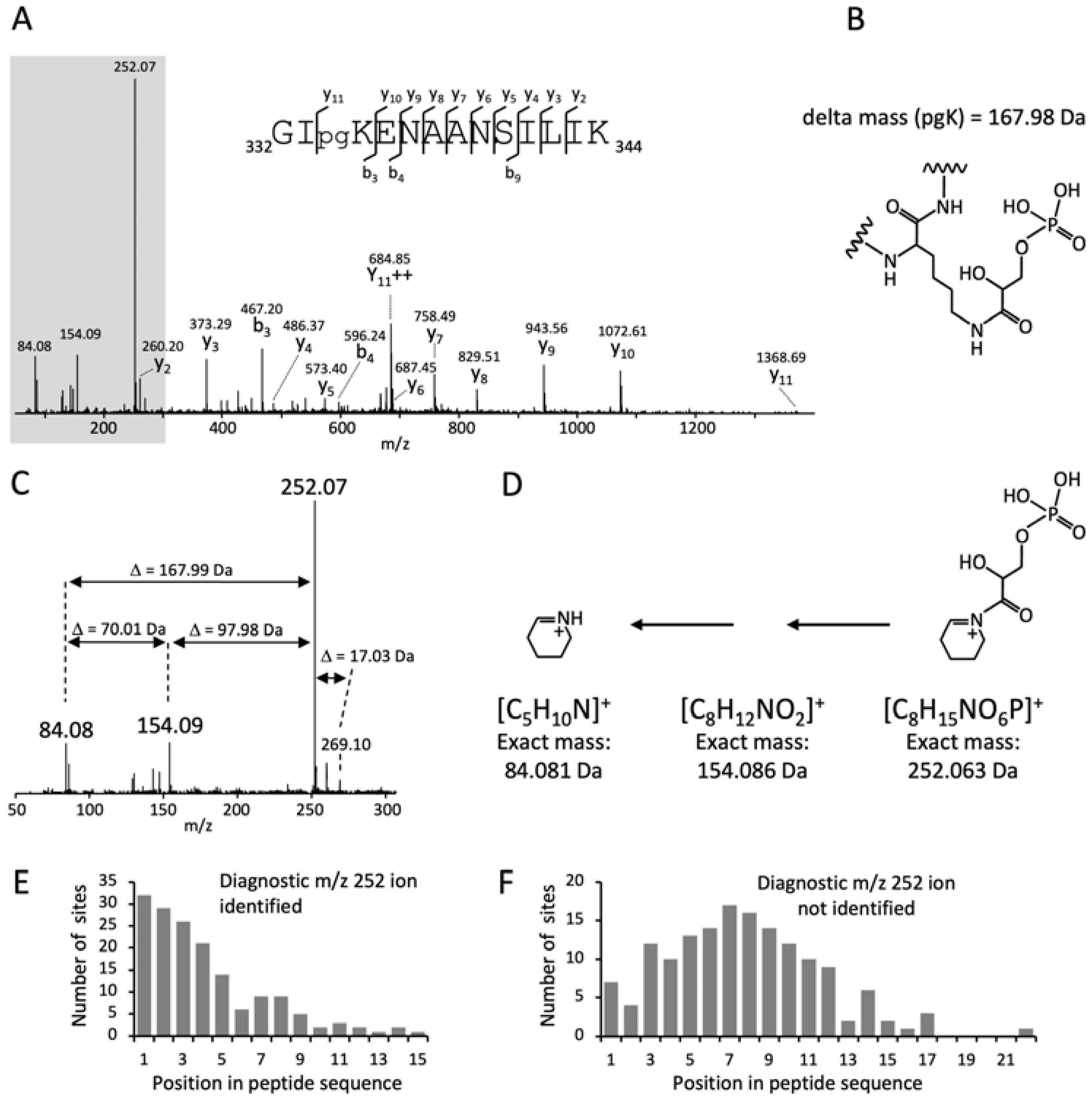
3.1. A Cyclic Immonium Ion of Phosphoglyceryl-Lysine Confirms Phosphoglycerylation
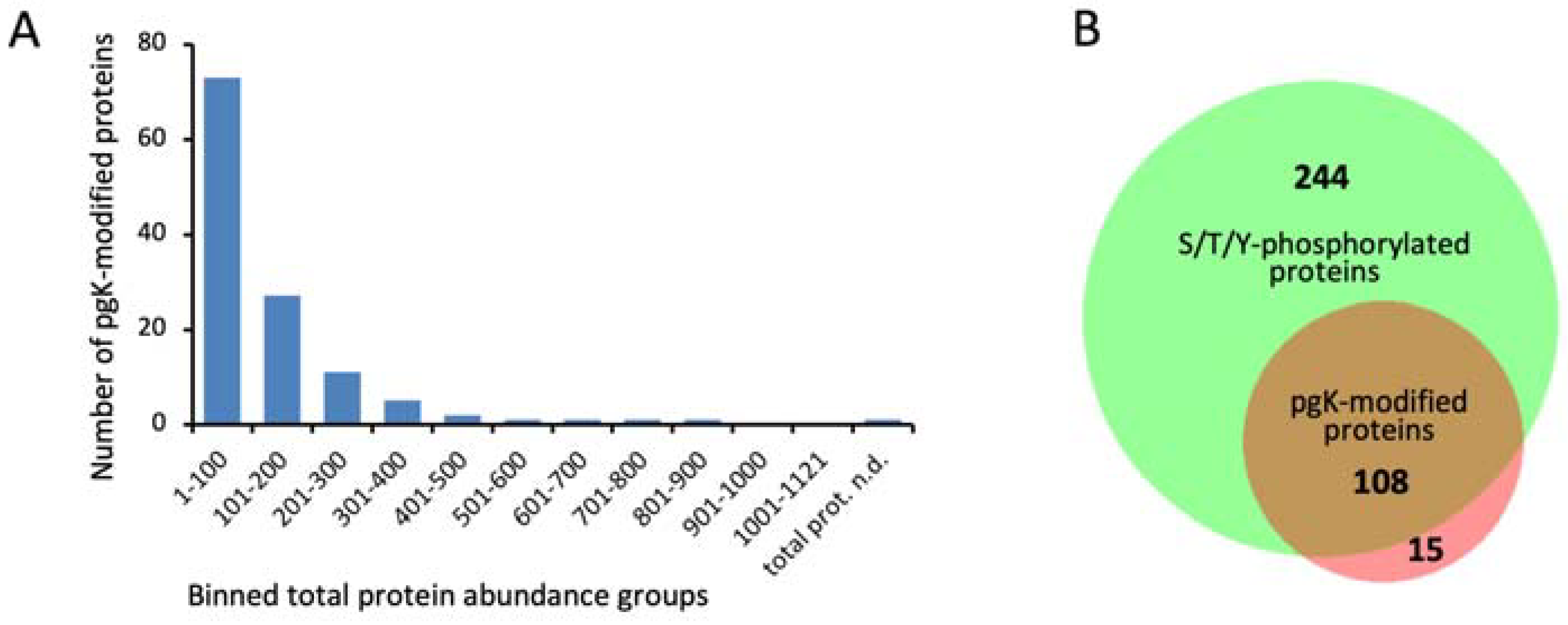
3.2. Identification of Phosphoglycerylation Depends on Protein Abundance
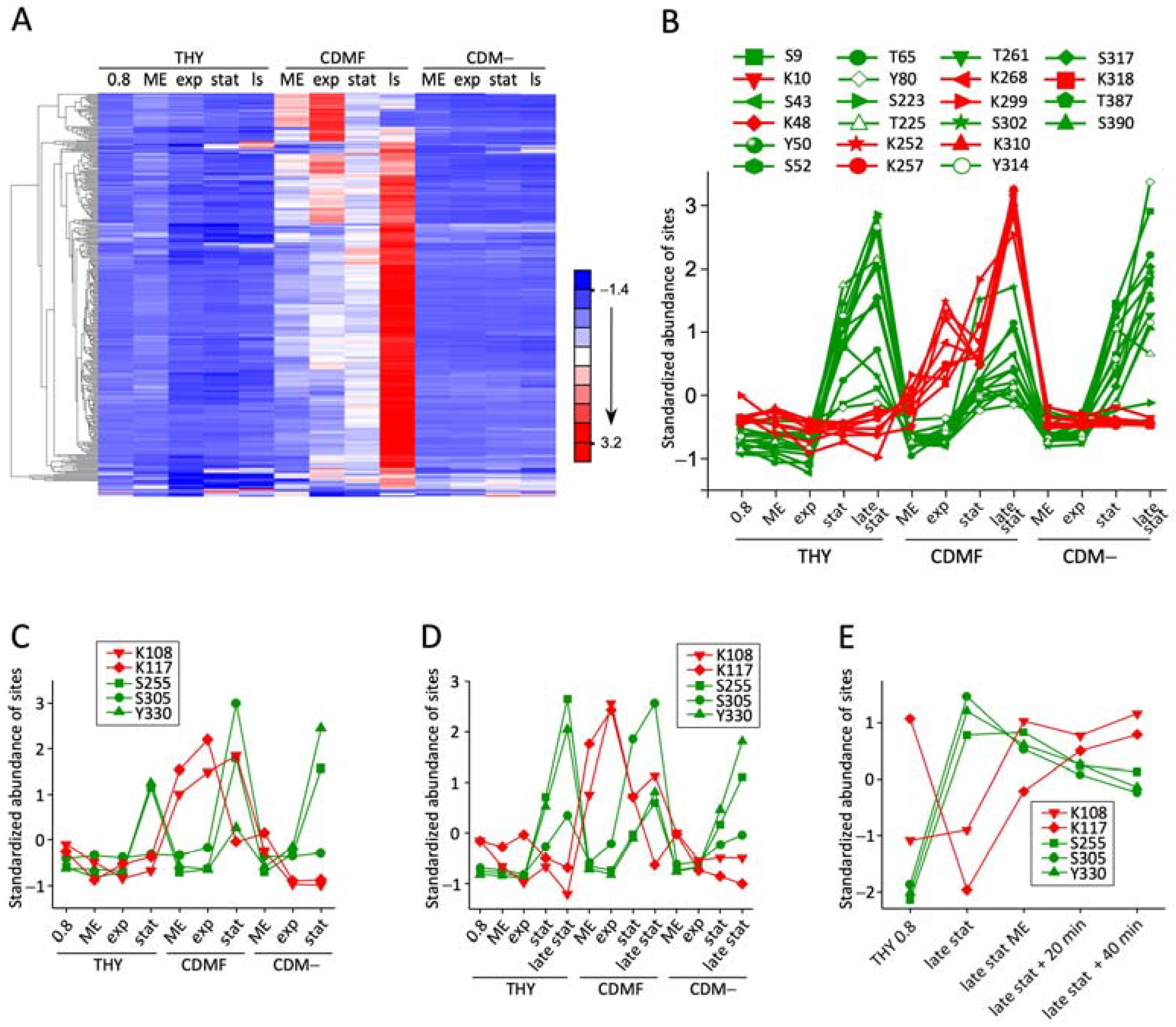
3.3. Phosphoglycerylation Accumulates during Growth with 1% Fructose
3.4. The Phosphoglyceryl Modification Is Mostly Low-Stoichiometric
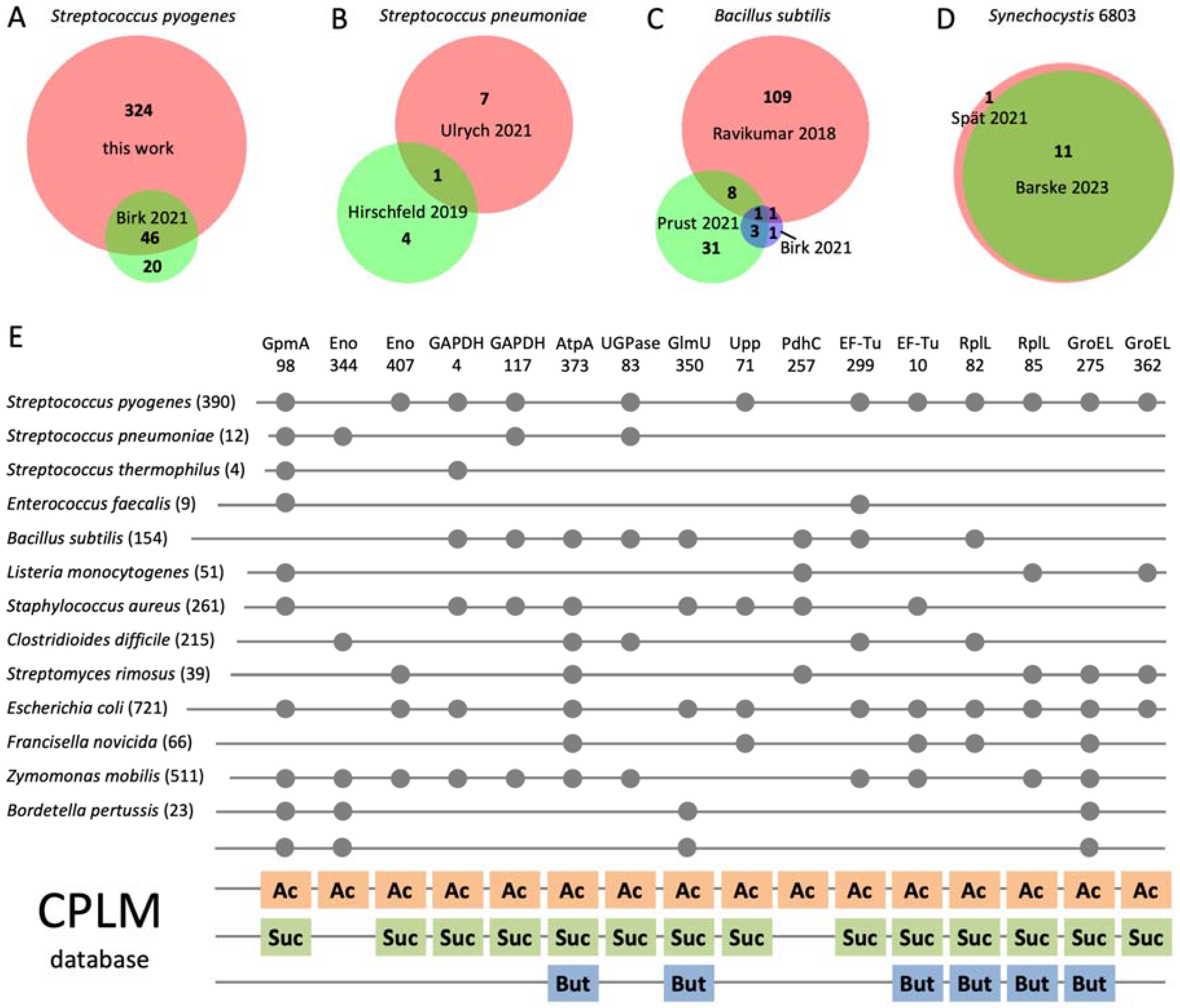
3.5. Phosphoglycerylation Is Conserved in Bacteria and Overlaps with Acylation
3.6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Macek, B.; Forchhammer, K.; Hardouin, J.; Weber-Ban, E.; Grangeasse, C.; Mijakovic, I. Protein post-translational modifications in bacteria. Nat. Reviews. Microbiol. 2019, 17, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.G.; Baumgartner, J.T.; Xie, X.; Jew, K.M.; Basisty, N.; Schilling, B.; Kuhn, M.L.; Wolfe, A.J. Mechanisms, Detection, and Relevance of Protein Acetylation in Prokaryotes. mBio 2019, 10, e02708-18. [Google Scholar] [CrossRef] [PubMed]
- Keenan, E.K.; Zachman, D.K.; Hirschey, M.D. Discovering the landscape of protein modifications. Mol. Cell 2021, 81, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, L.; Fu, Y.; Huo, M.; Qi, Q.; Zhao, G. Bacterial protein acetylation and its role in cellular physiology and metabolic regulation. Biotechnol. Adv. 2021, 53, 107842. [Google Scholar] [CrossRef] [PubMed]
- Nakayasu, E.S.; Burnet, M.C.; Walukiewicz, H.E.; Wilkins, C.S.; Shukla, A.K.; Brooks, S.; Plutz, M.J.; Lee, B.D.; Schilling, B.; Wolfe, A.J.; et al. Ancient Regulatory Role of Lysine Acetylation in Central Metabolism. mBio 2017, 8, e01894-17. [Google Scholar] [CrossRef] [PubMed]
- Popova, L.; Carr, R.A.; Carabetta, V.J. Recent Contributions of Proteomics to Our Understanding of Reversible N(epsilon)-Lysine Acylation in Bacteria. J. Proteome Res. 2024. [Google Scholar] [CrossRef] [PubMed]
- Boel, G.; Pichereau, V.; Mijakovic, I.; Maze, A.; Poncet, S.; Gillet, S.; Giard, J.C.; Hartke, A.; Auffray, Y.; Deutscher, J. Is 2-phosphoglycerate-dependent automodification of bacterial enolases implicated in their export? J. Mol. Biol. 2004, 337, 485–496. [Google Scholar] [CrossRef]
- Moellering, R.E.; Cravatt, B.F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 2013, 341, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.A.; Sharma, A.; Dehzangi, A.; Tsunoda, T. RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix. Genes 2020, 11, 1524. [Google Scholar] [CrossRef]
- Zhang, W.; Tan, X.; Lin, S.; Gou, Y.; Han, C.; Zhang, C.; Ning, W.; Wang, C.; Xue, Y. CPLM 4.0: An updated database with rich annotations for protein lysine modifications. Nucleic Acids Res. 2022, 50, D451–D459. [Google Scholar] [CrossRef]
- Molto, E.; Pintado, C.; Louzada, R.A.; Bernal-Mizrachi, E.; Andres, A.; Gallardo, N.; Bonzon-Kulichenko, E. Unbiased Phosphoproteome Mining Reveals New Functional Sites of Metabolite-Derived PTMs Involved in MASLD Development. Int. J. Mol. Sci. 2023, 24, 16172. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.; Sterck, L.; Dard, A.; Huang, J.; De Smet, I.; Gevaert, K.; Van Breusegem, F. The Plant PTM Viewer 2.0: In-depth exploration of plant protein modification landscapes. J. Exp. Bot. 2024, erae270. [Google Scholar] [CrossRef]
- Mikkat, S.; Kreutzer, M.; Patenge, N. Dynamic Protein Phosphorylation in Streptococcus pyogenes during Growth, Stationary Phase, and Starvation. Microorganisms 2024, 12, 621. [Google Scholar] [CrossRef]
- Kaufhold, A.; Podbielski, A.; Johnson, D.R.; Kaplan, E.L.; Lutticken, R. M protein gene typing of Streptococcus pyogenes by nonradioactively labeled oligonucleotide probes. J. Clin. Microbiol. 1992, 30, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- van de Rijn, I.; Kessler, R.E. Growth characteristics of group A streptococci in a new chemically defined medium. Infect. Immun. 1980, 27, 444–448. [Google Scholar] [CrossRef]
- Klahn, S.; Mikkat, S.; Riediger, M.; Georg, J.; Hess, W.R.; Hagemann, M. Integrative analysis of the salt stress response in cyanobacteria. Biol. Direct 2021, 16, 26. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Stucky, M.; Wakefield, C.; Melott, J.M.; Akbani, R.; Weinstein, J.N.; Broom, B.M. Interactive Clustered Heat Map Builder: An easy web-based tool for creating sophisticated clustered heat maps. F1000Research 2020, 8, 1750. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. The Clustal Omega Multiple Alignment Package. Methods Mol. Biol. 2021, 2231, 3–16. [Google Scholar] [CrossRef]
- Creasy, D.M.; Cottrell, J.S. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2002, 2, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Tholey, A.; Reed, J.; Lehmann, W.D. Electrospray tandem mass spectrometric studies of phosphopeptides and phosphopeptide analogues. J. Mass Spectrom. 1999, 34, 117–123. [Google Scholar] [CrossRef]
- Couttas, T.A.; Raftery, M.J.; Bernardini, G.; Wilkins, M.R. Immonium ion scanning for the discovery of post-translational modifications and its application to histones. J. Proteome Res. 2008, 7, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.W.; Schlosser, A.; Wei, J.; Lehmann, W.D. Collision-induced reporter fragmentations for identification of covalently modified peptides. Anal. Bioanal. Chem. 2007, 389, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Fenaille, F.; Tabet, J.C.; Guy, P.A. Study of peptides containing modified lysine residues by tandem mass spectrometry: Precursor ion scanning of hexanal-modified peptides. Rapid Commun. Mass Spectrom. 2004, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, T.; Harrison, A.G. Ion chemistry of protonated lysine derivatives. J. Mass Spectrom. 1996, 31, 1237–1243. [Google Scholar] [CrossRef]
- Muroski, J.M.; Fu, J.Y.; Nguyen, H.H.; Ogorzalek Loo, R.R.; Loo, J.A. Leveraging Immonium Ions for Targeting Acyl-Lysine Modifications in Proteomic Datasets. Proteomics 2021, 21, e2000111. [Google Scholar] [CrossRef] [PubMed]
- Muroski, J.M.; Fu, J.Y.; Nguyen, H.H.; Wofford, N.Q.; Mouttaki, H.; James, K.L.; McInerney, M.J.; Gunsalus, R.P.; Loo, J.A.; Ogorzalek Loo, R.R. The Acyl-Proteome of Syntrophus aciditrophicus Reveals Metabolic Relationships in Benzoate Degradation. Mol. Cell. Proteom. 2022, 21, 100215. [Google Scholar] [CrossRef]
- Trelle, M.B.; Jensen, O.N. Utility of immonium ions for assignment of epsilon-N-acetyllysine-containing peptides by tandem mass spectrometry. Anal. Chem. 2008, 80, 3422–3430. [Google Scholar] [CrossRef]
- Wan, N.; Wang, N.; Yu, S.; Zhang, H.; Tang, S.; Wang, D.; Lu, W.; Li, H.; Delafield, D.G.; Kong, Y.; et al. Cyclic immonium ion of lactyllysine reveals widespread lactylation in the human proteome. Nat. Methods 2022, 19, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Zolg, D.P.; Wilhelm, M.; Schmidt, T.; Medard, G.; Zerweck, J.; Knaute, T.; Wenschuh, H.; Reimer, U.; Schnatbaum, K.; Kuster, B. ProteomeTools: Systematic Characterization of 21 Post-translational Protein Modifications by Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Using Synthetic Peptides. Mol. Cell. Proteom. 2018, 17, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.F.; Yates, J.R., 3rd; Shabanowitz, J.; Winston, S.; Hauer, C.R. Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1986, 83, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Basisty, N.; Christensen, D.G.; Sorensen, D.; Orr, J.S.; Wolfe, A.J.; Rao, C.V. Global Lysine Acetylation in Escherichia coli Results from Growth Conditions That Favor Acetate Fermentation. J. Bacteriol. 2019, 201, e00768-18. [Google Scholar] [CrossRef] [PubMed]
- Birk, M.S.; Charpentier, E.; Frese, C.K. Automated Phosphopeptide Enrichment for Gram-Positive Bacteria. J. Proteome Res. 2021, 20, 4886–4892. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, C.; Gomez-Mejia, A.; Bartel, J.; Hentschker, C.; Rohde, M.; Maass, S.; Hammerschmidt, S.; Becher, D. Proteomic Investigation Uncovers Potential Targets and Target Sites of Pneumococcal Serine-Threonine Kinase StkP and Phosphatase PhpP. Front. Microbiol. 2019, 10, 3101. [Google Scholar] [CrossRef] [PubMed]
- Ulrych, A.; Fabrik, I.; Kupcik, R.; Vajrychova, M.; Doubravova, L.; Branny, P. Cell Wall Stress Stimulates the Activity of the Protein Kinase StkP of Streptococcus pneumoniae, Leading to Multiple Phosphorylation. J. Mol. Biol. 2021, 433, 167319. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, V.; Nalpas, N.C.; Anselm, V.; Krug, K.; Lenuzzi, M.; Sestak, M.S.; Domazet-Loso, T.; Mijakovic, I.; Macek, B. In-depth analysis of Bacillus subtilis proteome identifies new ORFs and traces the evolutionary history of modified proteins. Sci. Rep. 2018, 8, 17246. [Google Scholar] [CrossRef] [PubMed]
- Prust, N.; van der Laarse, S.; van den Toorn, H.W.P.; van Sorge, N.M.; Lemeer, S. In-Depth Characterization of the Staphylococcus aureus Phosphoproteome Reveals New Targets of Stk1. Mol. Cell. Proteom. 2021, 20, 100034. [Google Scholar] [CrossRef]
- Spat, P.; Barske, T.; Macek, B.; Hagemann, M. Alterations in the CO2 availability induce alterations in the phosphoproteome of the cyanobacterium Synechocystis sp. PCC 6803. New Phytol. 2021, 231, 1123–1137. [Google Scholar] [CrossRef]
- Barske, T.; Spat, P.; Schubert, H.; Walke, P.; Macek, B.; Hagemann, M. The Role of Serine/Threonine-Specific Protein Kinases in Cyanobacteria—SpkB Is Involved in Acclimation to Fluctuating Conditions in Synechocystis sp. PCC 6803. Mol. Cell. Proteom. 2023, 22, 100656. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Haller, L.; Blein-Nicolas, M.; Zivy, M.; Canette, A.; Verbrugghe, M.; Mezange, C.; Boulay, M.; Gardan, R.; Samson, S.; et al. Identification of Hanks-Type Kinase PknB-Specific Targets in the Streptococcus thermophilus Phosphoproteome. Front. Microbiol. 2019, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- Iannetta, A.A.; Minton, N.E.; Uitenbroek, A.A.; Little, J.L.; Stanton, C.R.; Kristich, C.J.; Hicks, L.M. IreK-Mediated, Cell Wall-Protective Phosphorylation in Enterococcus faecalis. J. Proteome Res. 2021, 20, 5131–5144. [Google Scholar] [CrossRef] [PubMed]
- Smits, W.K.; Mohammed, Y.; de Ru, A.H.; Cordo, V.; Friggen, A.H.; van Veelen, P.A.; Hensbergen, P.J. Clostridioides difficile Phosphoproteomics Shows an Expansion of Phosphorylated Proteins in Stationary Growth Phase. mSphere 2022, 7, e0091121. [Google Scholar] [CrossRef]
- Saric, E.; Quinn, G.A.; Nalpas, N.; Paradzik, T.; Kazazic, S.; Filic, Z.; Semanjski, M.; Herron, P.; Hunter, I.; Macek, B.; et al. Phosphoproteome Dynamics of Streptomyces rimosus during Submerged Growth and Antibiotic Production. mSystems 2022, 7, e0019922. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.M.; Lin, M.H.; Heck, A.J.R.; Lemeer, S. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat. Methods 2018, 15, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Ziveri, J.; Chhuon, C.; Jamet, A.; Rytter, H.; Prigent, G.; Tros, F.; Barel, M.; Coureuil, M.; Lays, C.; Henry, T.; et al. Critical Role of a Sheath Phosphorylation Site On the Assembly and Function of an Atypical Type VI Secretion System. Mol. Cell. Proteom. 2019, 18, 2418–2432. [Google Scholar] [CrossRef] [PubMed]
- Tatli, M.; Hebert, A.S.; Coon, J.J.; Amador-Noguez, D. Genome Wide Phosphoproteome Analysis of Zymomonas mobilis Under Anaerobic, Aerobic, and N(2)-Fixing Conditions. Front. Microbiol. 2019, 10, 1986. [Google Scholar] [CrossRef] [PubMed]
- Luu, L.D.W.; Zhong, L.; Kaur, S.; Raftery, M.J.; Lan, R. Comparative Phosphoproteomics of Classical Bordetellae Elucidates the Potential Role of Serine, Threonine and Tyrosine Phosphorylation in Bordetella Biology and Virulence. Front. Cell. Infect. Microbiol. 2021, 11, 660280. [Google Scholar] [CrossRef]
- Pearson, C.L.; Loshon, C.A.; Pedersen, L.B.; Setlow, B.; Setlow, P. Analysis of the function of a putative 2,3-diphosphoglyceric acid-dependent phosphoglycerate mutase from Bacillus subtilis. J. Bacteriol. 2000, 182, 4121–4123. [Google Scholar] [CrossRef]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, H.; Guo, Z.; Zou, S.; Long, F.; Wu, J.; Li, P.; Zhao, G.P.; Zhao, W. Global Insights Into Lysine Acylomes Reveal Crosstalk between Lysine Acetylation and Succinylation in Streptomyces coelicolor Metabolic Pathways. Mol. Cell. Proteom. 2021, 20, 100148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, T.; Wang, L.; Huang, Y.; Fan, R.; Ma, K.; Kan, Y.; Tan, M.; Xu, J.Y. Global landscape of lysine acylomes in Bacillus subtilis. J. Proteom. 2023, 271, 104767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Dong, Q.; Zhu, J.; Peng, R.; He, C.; Li, Y.; Lin, R.; Jiang, P.; Zheng, M.; et al. Lysine Acylation Modification Landscape of Brucella abortus Proteome and its Virulent Proteins. Front. Cell Dev. Biol. 2022, 10, 839822. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Yang, M.; Chen, Z.; Cheng, Z.; Yi, X.; Li, C.; He, C.; Xiong, Q.; Chen, H.; Wang, Q.; et al. Acetylome analysis reveals the involvement of lysine acetylation in photosynthesis and carbon metabolism in the model cyanobacterium Synechocystis sp. PCC 6803. J. Proteome Res. 2015, 14, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Nisar, A.; Gongye, X.; Huang, Y.; Khan, S.; Chen, M.; Wu, B.; He, M. Genome-Wide Analyses of Proteome and Acetylome in Zymomonas mobilis Under N2-Fixing Condition. Front. Microbiol. 2021, 12, 740555. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Xu, Z.; Liu, X.; Tan, M.; Ye, B.C. Protein Acetylation and Butyrylation Regulate the Phenotype and Metabolic Shifts of the Endospore-forming Clostridium acetobutylicum. Mol. Cell. Proteom. 2018, 17, 1156–1169. [Google Scholar] [CrossRef]
- Brunk, E.; Chang, R.L.; Xia, J.; Hefzi, H.; Yurkovich, J.T.; Kim, D.; Buckmiller, E.; Wang, H.H.; Cho, B.K.; Yang, C.; et al. Characterizing posttranslational modifications in prokaryotic metabolism using a multiscale workflow. Proc. Natl. Acad. Sci. USA 2018, 115, 11096–11101. [Google Scholar] [CrossRef]
- Fatema, N.; Li, X.; Gan, Q.; Fan, C. Characterizing lysine acetylation of glucokinase. Protein Sci. 2024, 33, e4845. [Google Scholar] [CrossRef]
- Schastnaya, E.; Doubleday, P.F.; Maurer, L.; Sauer, U. Non-enzymatic acetylation inhibits glycolytic enzymes in Escherichia coli. Cell Rep. 2023, 42, 111950. [Google Scholar] [CrossRef]
- Venkat, S.; Chen, H.; Stahman, A.; Hudson, D.; McGuire, P.; Gan, Q.; Fan, C. Characterizing Lysine Acetylation of Isocitrate Dehydrogenase in Escherichia coli. J. Mol. Biol. 2018, 430, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikkat, S.; Kreutzer, M.; Patenge, N. Lysine Phoshoglycerylation Is Widespread in Bacteria and Overlaps with Acylation. Microorganisms 2024, 12, 1556. https://doi.org/10.3390/microorganisms12081556
Mikkat S, Kreutzer M, Patenge N. Lysine Phoshoglycerylation Is Widespread in Bacteria and Overlaps with Acylation. Microorganisms. 2024; 12(8):1556. https://doi.org/10.3390/microorganisms12081556
Chicago/Turabian StyleMikkat, Stefan, Michael Kreutzer, and Nadja Patenge. 2024. "Lysine Phoshoglycerylation Is Widespread in Bacteria and Overlaps with Acylation" Microorganisms 12, no. 8: 1556. https://doi.org/10.3390/microorganisms12081556