
Breaking the Cycle: A Yeast Mannan-Rich Fraction Beneficially Modulates Egg Quality and the Antimicrobial Resistome Associated with Layer Hen Caecal Microbiomes under Commercial Conditions
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Design, Sample Collection and Preservation
2.3. Performance Data
2.4. DNA Extraction, Library Construction, Quality Control, and Sequencing
2.5. Data Reprocessing and Assembly
2.6. Gene Prediction and Abundance Analysis
2.7. Species Annotation
2.8. Resistance Gene Annotation
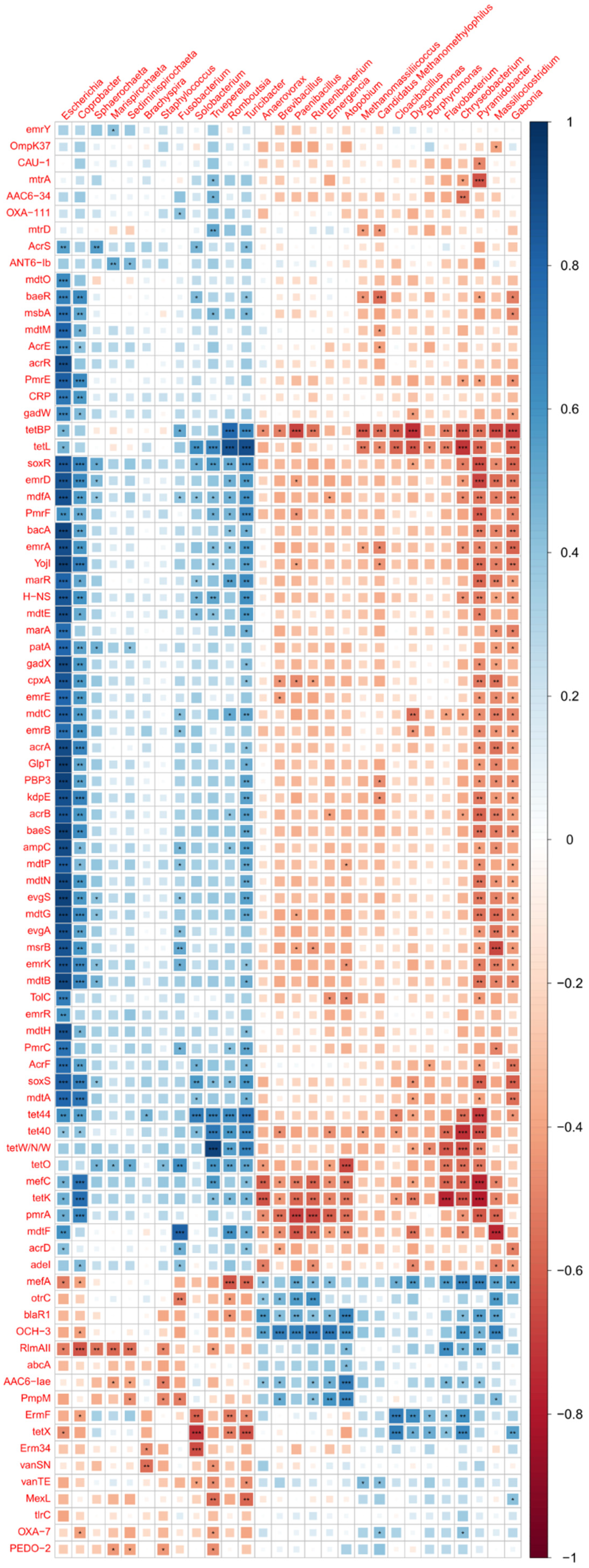
2.9. Bacterial Composition vs. Antimicrobial Resistance Genes
3. Results
3.1. Performance
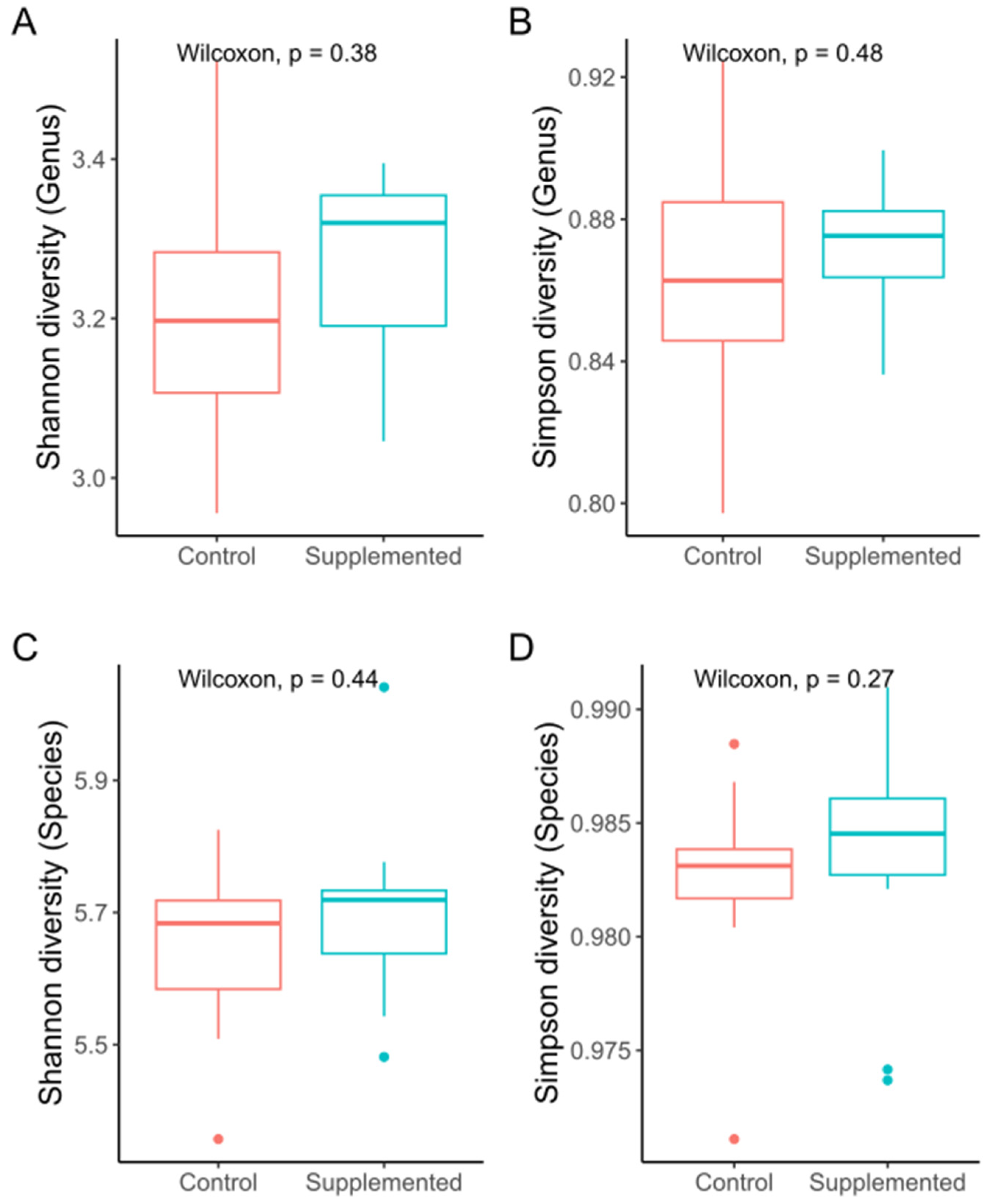
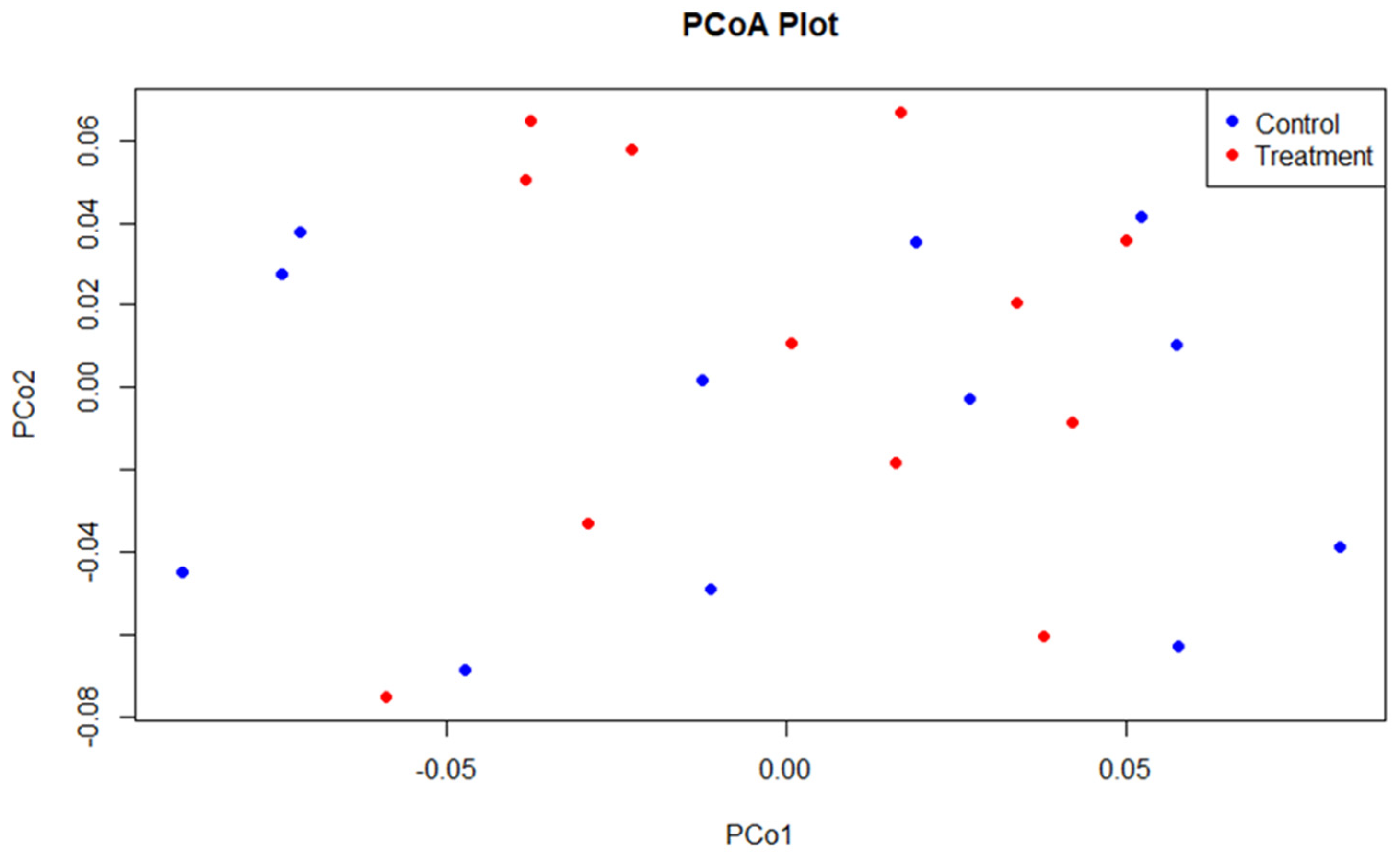
3.2. Sequencing and Microbial Diversity
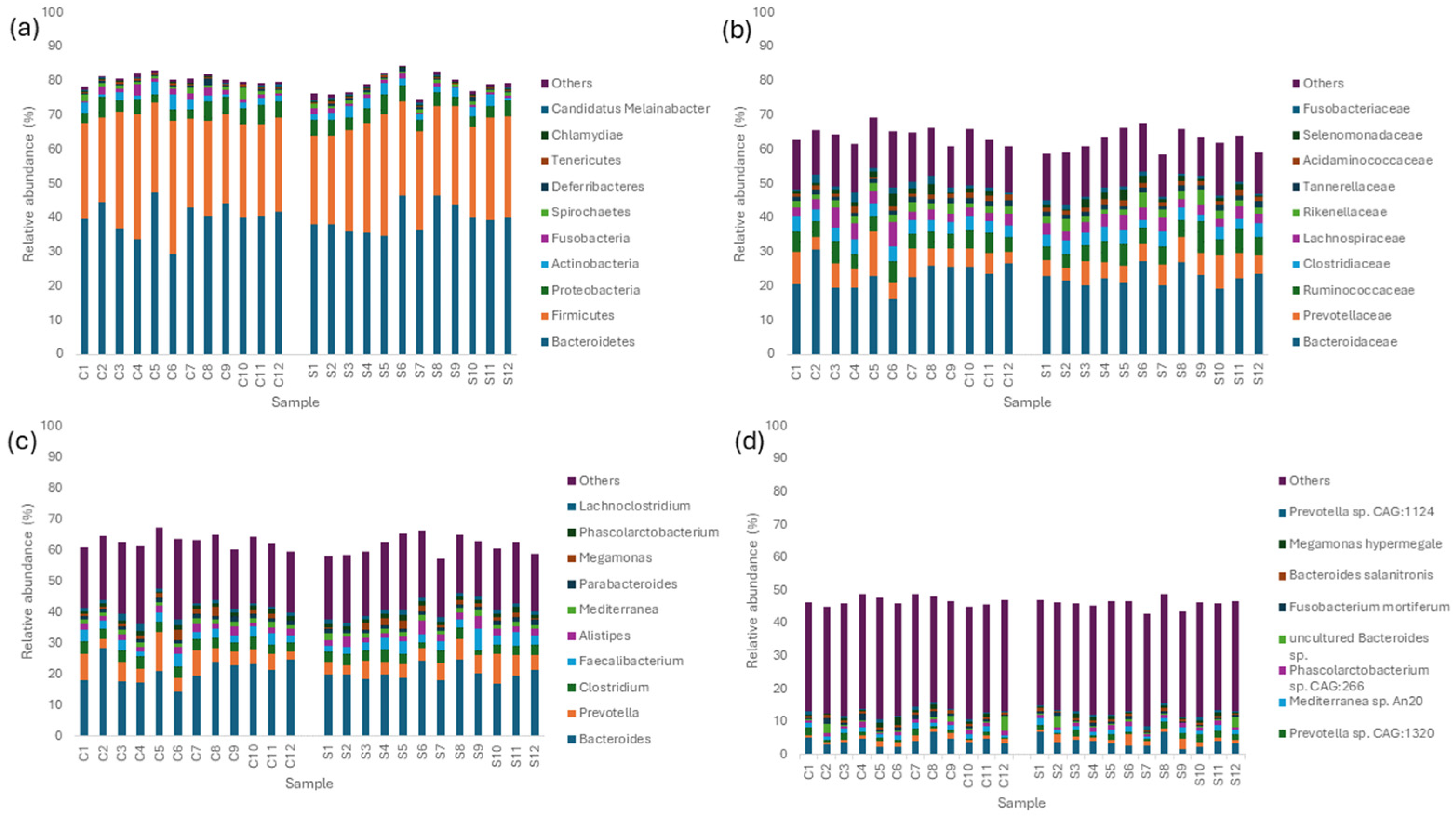
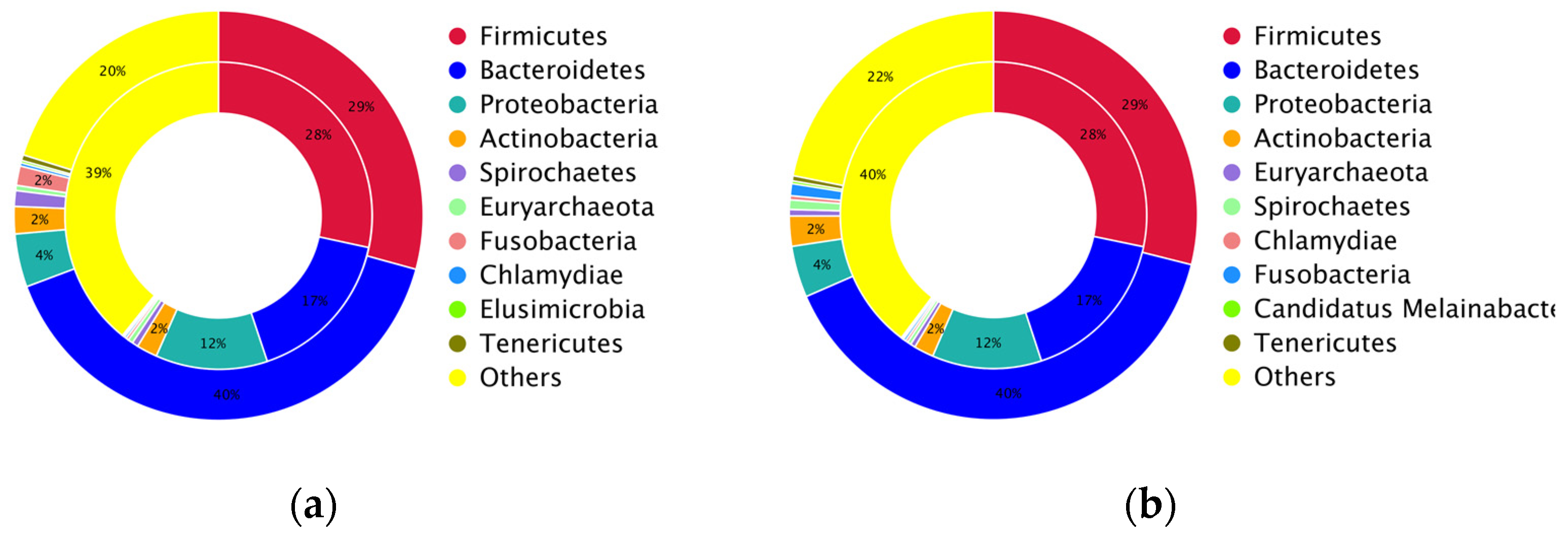
3.3. Taxonomic Abundance and Composition of the Layer Caecal Microbiota
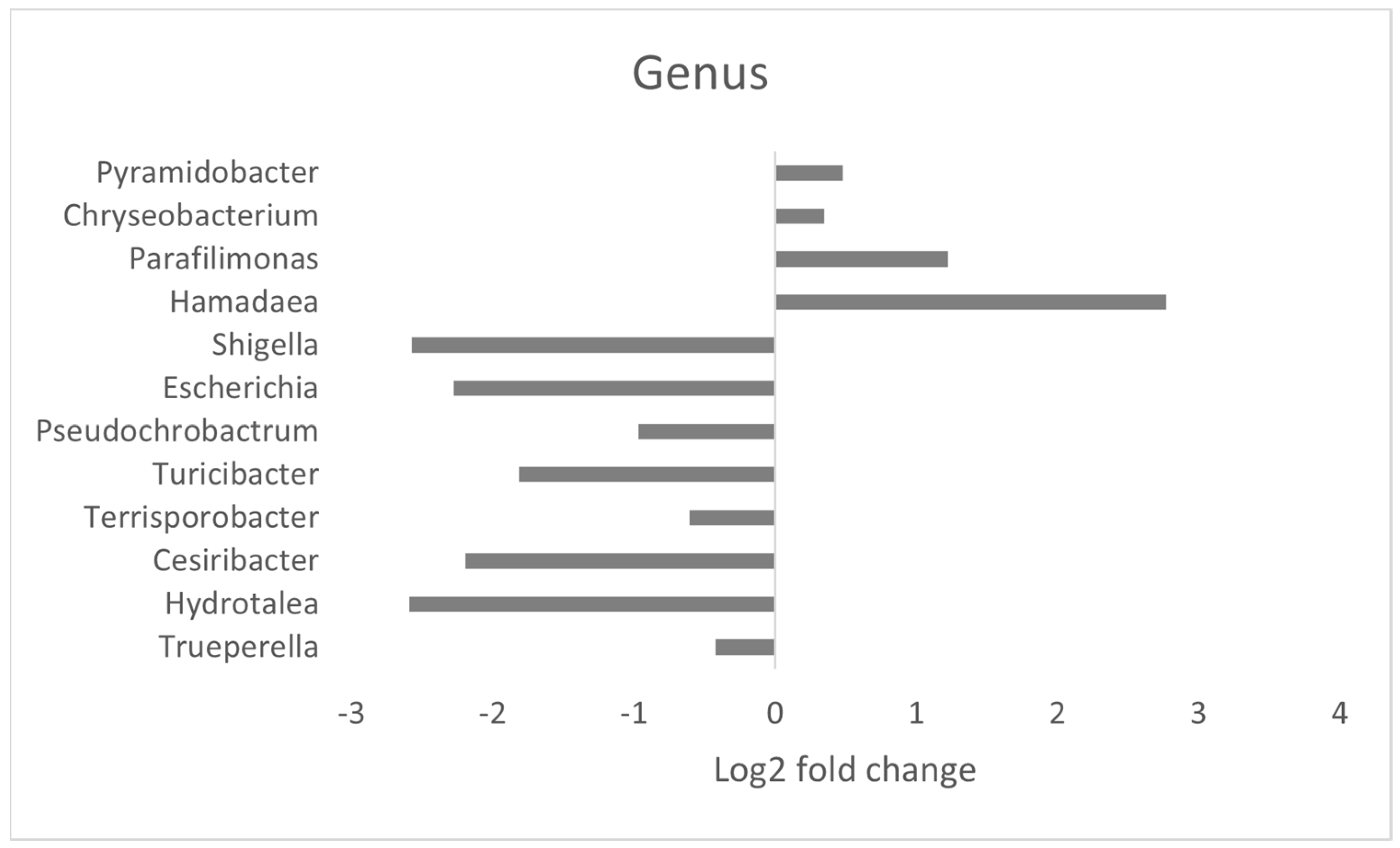
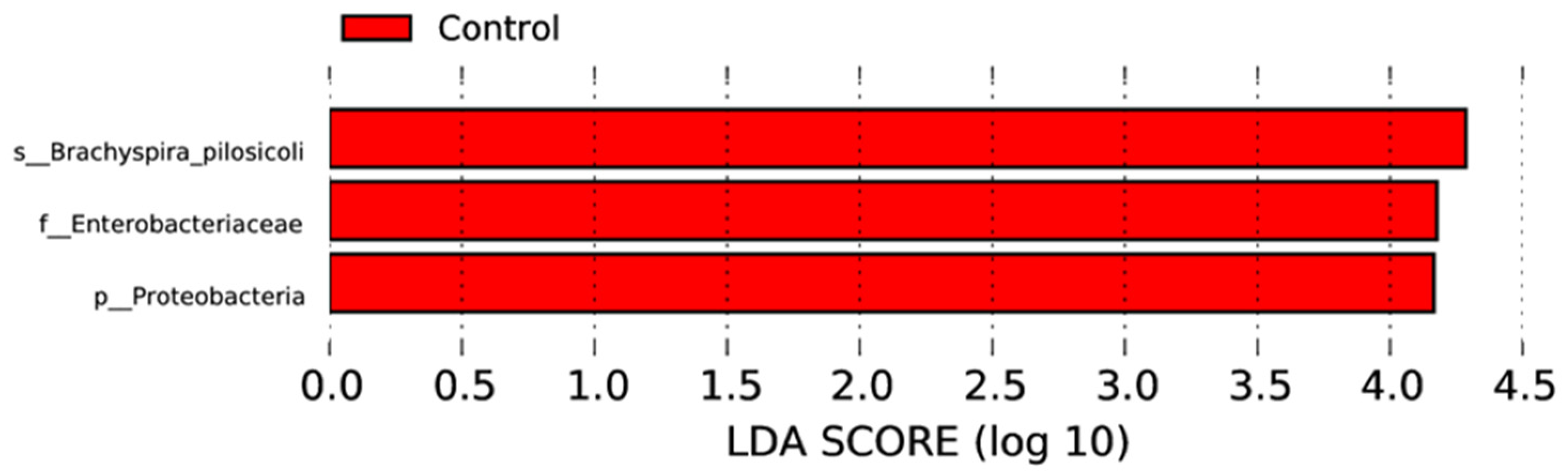
3.4. Effect of MRF Supplementation on Bacterial Pathobionts
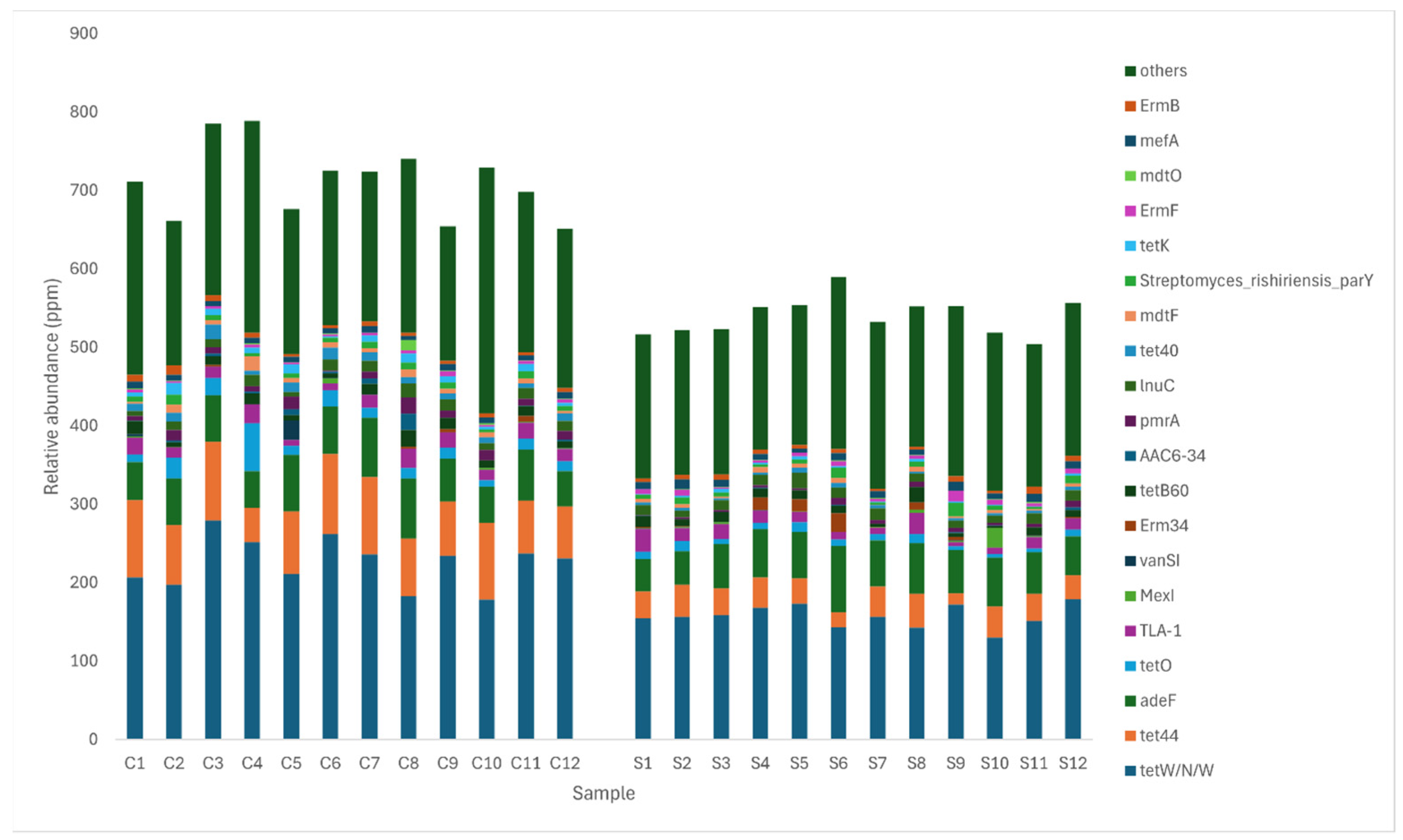
3.5. Comparison of Antibiotic Resistance Genes between Control and MRF Supplemented Layers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wickramasuriya, S.S.; Park, I.; Lee, K.; Lee, Y.; Kim, W.H.; Nam, H.; Lillehoj, H.S. Role of Physiology, Immunity, Microbiota, and Infectious Diseases in the Gut Health of Poultry. Vaccines 2022, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Aruwa, C.E.; Pillay, C.; Nyaga, M.M.; Sabiu, S. Poultry gut health–microbiome functions, environmental impacts, microbiome engineering and advancements in characterization technologies. J. Anim. Sci. Biotechnol. 2021, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Diaz Carrasco, J.M.; Casanova, N.A.; Fernández Miyakawa, M.E. Microbiota, Gut Health and Chicken Productivity: What Is the Connection? Microorganisms 2019, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.; Semedo-Lemsaddek, T.; Cunha, E.; Tavares, L.; Oliveira, M. Antimicrobial Drug Resistance in Poultry Production: Current Status and Innovative Strategies for Bacterial Control. Microorganisms 2023, 11, 953. [Google Scholar] [CrossRef] [PubMed]
- Koorakula, R.; Schiavinato, M.; Ghanbari, M.; Wegl, G.; Grabner, N.; Koestelbauer, A.; Klose, V.; Dohm, J.C.; Domig, K.J. Metatranscriptomic Analysis of the Chicken Gut Resistome Response to In-Feed Antibiotics and Natural Feed Additives. Front. Microbiol. 2022, 13, 833790. [Google Scholar] [CrossRef]
- Rahman, M.R.T.; Fliss, I.; Biron, E. Insights in the Development and Uses of Alternatives to Antibiotic Growth Promoters in Poultry and Swine Production. Antibiotics 2022, 11, 766. [Google Scholar] [CrossRef]
- Salami, S.A.; Ross, S.A.; Patsiogiannis, A.; Moran, C.A.; Taylor-Pickard, J. Performance and environmental impact of egg production in response to dietary supplementation of mannan oligosaccharide in laying hens: A meta-analysis. Poult. Sci. 2022, 101, 101745. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, A.; Leigh, R.J.; Walsh, F.; Murphy, R. Microbial community diversity and structure in the cecum of laying hens with and without mannan-rich fraction supplementation. J. Appl. Poult. Res. 2023, 32, 100342. [Google Scholar] [CrossRef]
- Leigh, R.J.; Corrigan, A.; Murphy, R.A.; Walsh, F. Yeast mannan rich fraction positively influences microbiome uniformity, productivity associated taxa, and lay performance. Anim. Microbiome 2024, 6, 9. [Google Scholar] [CrossRef]
- Grant, S.; Smith, H.; Murphy, R. Mannan based prebiotics modulate growth rate and energy phenotype of tetracycline resistant E. coli. Front. Anim. Sci. 2023, 3, 1069280. [Google Scholar] [CrossRef]
- Smith, H.; Grant, S.; Parker, J.; Murphy, R. Yeast cell wall mannan rich fraction modulates bacterial cellular respiration potentiating antibiotic efficacy. Sci. Rep. 2020, 10, 21880. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Grant, S.; Meleady, P.; Henry, M.; O’Gorman, D.; Clynes, M.; Murphy, R. Yeast Mannan-Rich Fraction Modulates Endogenous Reactive Oxygen Species Generation and Antibiotic Sensitivity in Resistant E. coli. Int. J. Mol. Sci. 2022, 24, 218. [Google Scholar] [CrossRef]
- Parker, J.; Smith, H.; Corrigan, A.; Fay, B.J.; Murphy, R. Effects of dietary mannan-rich fraction on microbial populations in broiler chickens. In Proceedings of the 6th International Conference on Poultry Intestinal Health, Rome, Italy, 3–5 April 2019. [Google Scholar]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLOS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Barnabas, B.; Blakesley, R.; Bouffard, G.; Brooks, S.; Coleman, H.; Dekhtyar, M.; et al. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Villar, E.; Farrant, G.K.; Follows, M.; Garczarek, L.; Speich, S.; Audic, S.; Bittner, L.; Blanke, B.; Brum, J.R.; Brunet, C.; et al. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science 2015, 348, 1261447. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Avershina, E.; Frisli, T.; Rudi, K. De novo semi-alignment of 16S rRNA gene sequences for deep phylogenetic characterization of next generation sequencing data. Microbes Environ. 2013, 28, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Noval Rivas, M.; Burton, O.T.; Wise, P.; Zhang, Y.Q.; Hobson, S.A.; Garcia Lloret, M.; Chehoud, C.; Kuczynski, J.; DeSantis, T.; Warrington, J.; et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 201–212. [Google Scholar] [CrossRef]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLOS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, D566–D573. [Google Scholar] [CrossRef]
- Hooge, D. Meta-analysis of Broiler Chicken Pen Trials Evaluating Dietary Mannan Oligosaccharide, 1993–2003. Int. J. Poult. Sci. 2004, 3, 163–174. [Google Scholar]
- Dilawar, M.A.; Mun, H.S.; Rathnayake, D.; Yang, E.J.; Seo, Y.S.; Park, H.S.; Yang, C.J. Egg Quality Parameters, Production Performance and Immunity of Laying Hens Supplemented with Plant Extracts. Animals 2021, 11, 975. [Google Scholar] [CrossRef]
- Corrigan, A.; de Leeuw, M.; Penaud-Frézet, S.; Dimova, D.; Murphy, R.A. Phylogenetic and functional alterations in bacterial community compositions in broiler ceca as a result of mannan oligosaccharide supplementation. Appl. Environ. Microbiol. 2015, 81, 3460–3470. [Google Scholar] [CrossRef]
- Corrigan, A.; Fay, B.J.; Corcionivoschi, N.; Murphy, R.A. Effect of yeast mannan-rich fractions on reducing Campylobacter colonization in broiler chickens. J. Appl. Poult. Res. 2017, 26, 350–357. [Google Scholar] [CrossRef]
- Corrigan, A.; Russell, N.; Welge, M.; Auvil, L.; Bushell, C.; White, B.A.; Murphy, R.A. The use of random forests modelling to detect yeast-mannan sensitive bacterial changes in the broiler cecum. Sci. Rep. 2018, 8, 13270. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, C.; Corrigan, A.; Moynagh, P.; Murphy, R. Effect of yeast cell wall supplementation on intestinal integrity, digestive enzyme activity and immune traits of broilers. Br. Poult. Sci. 2021, 62, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Joat, N.; Van, T.T.H.; Stanley, D.; Moore, R.J.; Chousalkar, K. Temporal dynamics of gut microbiota in caged laying hens: A field observation from hatching to end of lay. Appl. Microbiol. Biotechnol. 2021, 105, 4719–4730. [Google Scholar] [CrossRef]
- Wei, J.; Xiao, H.; Wei, Y.; Tsopmejio, I.S.N.; Sun, C.; Wu, H.; Jin, Z.; Song, H. Longitudinal Study of the Effects of Flammulina velutipes Stipe Wastes on the Cecal Microbiota of Laying Hens. mSystems 2023, 8, e00835-22. [Google Scholar] [CrossRef] [PubMed]
- Mon, K.K.; Saelao, P.; Halstead, M.M.; Chanthavixay, G.; Chang, H.C.; Garas, L.; Maga, E.A.; Zhou, H. Salmonella enterica Serovars Enteritidis Infection Alters the Indigenous Microbiota Diversity in Young Layer Chicks. Front. Vet. Sci. 2015, 2, 61. [Google Scholar] [CrossRef]
- Dai, D.; Qi, G.-h.; Wang, J.; Zhang, H.-j.; Qiu, K.; Wu, S.-g. Intestinal microbiota of layer hens and its association with egg quality and safety. Poult. Sci. 2022, 101, 102008. [Google Scholar] [CrossRef]
- Videnska, P.; Sedlar, K.; Lukac, M.; Faldynova, M.; Gerzova, L.; Cejkova, D.; Sisak, F.; Rychlik, I. Succession and Replacement of Bacterial Populations in the Caecum of Egg Laying Hens over Their Whole Life. PLoS ONE 2014, 9, e115142. [Google Scholar] [CrossRef]
- Van Goor, A.; Redweik, G.A.J.; Stromberg, Z.R.; Treadwell, C.G.; Xin, H.; Mellata, M. Microbiome and biological blood marker changes in hens at different laying stages in conventional and cage free housings. Poult. Sci. 2020, 99, 2362–2374. [Google Scholar] [CrossRef]
- Xu, H.; Lu, Y.; Li, D.; Yan, C.; Jiang, Y.; Hu, Z.; Zhang, Z.; Du, R.; Zhao, X.; Zhang, Y.; et al. Probiotic mediated intestinal microbiota and improved performance, egg quality and ovarian immune function of laying hens at different laying stage. Front. Microbiol. 2023, 14, 1041072. [Google Scholar] [CrossRef]
- Leigh, R.J.; Corrigan, A.; Murphy, R.A.; Walsh, F. Effect of Mannan-rich fraction supplementation on commercial broiler intestinum tenue and cecum microbiota. Anim. Microbiome 2022, 4, 66. [Google Scholar] [CrossRef]
- Spring, P.; Wenk, C.; Connolly, A.; Kiers, A. A review of 733 published trials on Bio-Mos®, a mannan oligosaccharide, and Actigen®, a second generation mannose rich fraction, on farm and companion animals. J. Appl. Anim. Nutr. 2015, 3, e8. [Google Scholar] [CrossRef]
- Rzewuska, M.; Kwiecień, E.; Chrobak-Chmiel, D.; Kizerwetter-Świda, M.; Stefańska, I.; Gieryńska, M. Pathogenicity and Virulence of Trueperella pyogenes: A Review. Int. J. Mol. Sci. 2019, 20, 2737. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Liu, M.; Zhang, X.; Wang, H.; Yue, B. In vitro and in vivo expression of virulence genes in Trueperella pyogenes based on a mouse model. Vet. Microbiol. 2013, 163, 344–350. [Google Scholar] [CrossRef]
- Thanassi David, G.; Nuccio, S.-P.; Shu Kin So, S.; Bäumler Andreas, J. Fimbriae: Classification and Biochemistry. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef]
- Passey, J.L.; La Ragione, R.M. JMM Profile: Brachyspira species: The causative agent of Avian Intestinal Spirochaetosis. J. Med. Microbiol. 2022, 71, 001495. [Google Scholar] [CrossRef] [PubMed]
- Rebollada-Merino, A.; Ugarte-Ruiz, M.; Gómez-Buendía, A.; Bárcena, C.; García, N.; Domínguez, L.; Rodríguez-Bertos, A. Cecal Reduction of Brachyspira and Lesion Severity in Laying Hens Supplemented with Fermented Defatted ‘Alperujo’. Fermentation 2022, 8, 550. [Google Scholar] [CrossRef]
- Leigh, R.J.; Murphy, R.A.; Walsh, F. uniForest: An unsupervised machine learning technique to detect outliers and restrict variance in microbiome studies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Glendinning, L.; McLachlan, G.; Vervelde, L. Age-related differences in the respiratory microbiota of chickens. PLoS ONE 2017, 12, e0188455. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Baranowski, E.; Nouvel, L.X.; Citti, C. The Airway Pathobiome in Complex Respiratory Diseases: A Perspective in Domestic Animals. Front. Cell. Infect. Microbiol. 2021, 11, 583600. [Google Scholar] [CrossRef]
- Kursa, O.; Tomczyk, G.; Sieczkowska, A.; Sawicka-Durkalec, A. Prevalence, Identification and Antibiotic Resistance of Gallibacterium anatis Isolates from Chickens in Poland. Pathogens 2023, 12, 992. [Google Scholar] [CrossRef]
- Michiels, T.; Welby, S.; Vanrobaeys, M.; Quinet, C.; Rouffaer, L.; Lens, L.; Martel, A.; Butaye, P. Prevalence of Mycoplasma gallisepticum and Mycoplasma synoviae in commercial poultry, racing pigeons and wild birds in Belgium. Avian Pathol. 2016, 45, 244–252. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Guerra, A.; Baker, M.; Hu, Y.; Wang, W.; Zhang, X.; Rong, J.; Zhang, Y.; Zhang, J.; Kaler, J.; Renney, D.; et al. Dissecting microbial communities and resistomes for interconnected humans, soil, and livestock. ISME J. 2023, 17, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lyu, N.; Liu, F.; Liu, W.J.; Bi, Y.; Zhang, Z.; Ma, S.; Cao, J.; Song, X.; Wang, A.; et al. More diversified antibiotic resistance genes in chickens and workers of the live poultry markets. Environ. Int. 2021, 153, 106534. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Wang, Y.; Sun, Y.; Ma, L.; Zeng, Q.; Jiang, X.; Li, A.; Zeng, Z.; Zhang, T. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Moore-Machacek, A.s.; Gloe, A.; O’Leary, N.; Reen, F.J. Efflux, Signaling and Warfare in a Polymicrobial World. Antibiotics 2023, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.; Fasolino, T.; Ivankovic, D.; Davis, N.J.; Brownlee, N. Genetic Factors That Contribute to Antibiotic Resistance through Intrinsic and Acquired Bacterial Genes in Urinary Tract Infections. Microorganisms 2023, 11, 1407. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.; McCarthy, C.G.P.; Murphy, S.; Walsh, F. Antibiotic resistomes of healthy pig faecal metagenomes. Microb. Genom. 2019, 5, e000272. [Google Scholar] [CrossRef] [PubMed]
- Browne, N.; Traynor, A.; Horgan, K.A. Mannan rich fraction from yeast modulates inflammatory responses in intestinal cells (HT-29) exposed to Escherichia coli. J. Appl. Anim. Nutr. 2019, 7, e5. [Google Scholar] [CrossRef]
- Almansour, A.M.; Alhadlaq, M.A.; Alzahrani, K.O.; Mukhtar, L.E.; Alharbi, A.L.; Alajel, S.M. The Silent Threat: Antimicrobial-Resistant Pathogens in Food-Producing Animals and Their Impact on Public Health. Microorganisms 2023, 11, 2127. [Google Scholar] [CrossRef]
- Yang, J.; Tong, C.; Xiao, D.; Xie, L.; Zhao, R.; Huo, Z.; Tang, Z.; Hao, J.; Zeng, Z.; Xiong, W. Metagenomic Insights into Chicken Gut Antibiotic Resistomes and Microbiomes. Microbiol. Spectr. 2022, 10, e01907–e01921. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, S.; Shajahan, S.; Prathiush, P.R.; Anjana, V.M.; Viswanathan, A.; Chandran, V.; Ajith Kumar, G.S.; Jayachandran, R.; Mathew, J.; Radhakrishnan, E.K. Healthy broilers disseminate antibiotic resistance in response to tetracycline input in feed concentrates. Microb. Pathog. 2020, 149, 104562. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W24 | W30 | W38 | W45 | W52 | W59 | W63 | W70 | W75 | W24-75 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Eggshell strength (kg.f) | Control | 5.49 | 5.09 a | 4.92 a | 4.51 a | 4.43 | 4.54 a | 3.96 a | 4.4 | 3.87 a | 4.55 a |
| Supplemented | 5.59 | 5.920 b | 5.24 b | 5.28 b | 4.88 | 5.13 b | 5.00 b | 4.52 | 4.33 b | 5.10 b | |
| W20-29 | W30-39 | W40-49 | W50-59 | W60-69 | W70-76 | W20-76 | |||||
| ADFI | Control | 115.8 | 127.3 | 143.6 | 146.4 | 125.3 | 133 | 130.2 | |||
| Supplemented | 117.6 | 140.2 | 133.2 | 129.0 | 123.5 | 115.6 | 127.2 | ||||
| Mortality | Control | 0.019 | 0.044 | 0.013 | 0.089 | 0.271 | 0.267 | 0.063 | |||
| Supplemented | 0.019 | 0.013 | 0.032 | 0.057 | 0.221 | 0.288 | 0.051 | ||||
| Second-grade eggs | Control | 4.45 | 4.95 a | 5.95 a | 8.25 a | 10.27 | 10.97 a | 6.51 | |||
| Supplemented | 4.36 | 4.40 b | 5.16 b | 7.48 b | 8.81 | 12.39 b | 6.05 |
| Drug Class | Resistance Mechanism | ARO Name | Control Mean (%) | Supplemented Mean (%) | Log2 Fold Change | Control Genome | Supplemented Genome | Phylogenetic Association |
|---|---|---|---|---|---|---|---|---|
| fluoroquinolone antibiotic; cephalosporin; glycylcycline; penam; tetracycline antibiotic; rifamycin antibiotic; phenicol antibiotic; triclosan | antibiotic target alteration; antibiotic efflux | Escherichia_coli_soxR | 1.07 × 10−06 | 5.37 × 10−08 | −4.31 | Core | Acc | Proteobacteria |
| fluoroquinolone antibiotic; cephalosporin; glycylcycline; penam; tetracycline antibiotic; rifamycin antibiotic; phenicol antibiotic; triclosan | antibiotic target alteration; antibiotic efflux | Escherichia_coli_marR | 8.34 × 10−07 | 7.18 × 10−08 | −3.54 | Acc | Acc | Bacteria |
| macrolide antibiotic | antibiotic efflux | mefC | 2.19 × 10−06 | 2.16 × 10−07 | −3.35 | Core | Acc | Bacteroidetes; Bacteroides coprophilus CAG:333 |
| fluoroquinolone antibiotic | antibiotic efflux | emrA | 8.90 × 10−07 | 8.90 × 10−08 | −3.32 | Acc | Acc | Proteobacteria |
| aminoglycoside antibiotic | antibiotic efflux | kdpE | 8.52 × 10−07 | 8.74 × 10−08 | −3.28 | Acc | Acc | Proteobacteria |
| aminoglycoside antibiotic; aminocoumarin antibiotic | antibiotic efflux | baeS | 7.56 × 10−07 | 8.62 × 10−08 | −3.13 | Core | Acc | Proteobacteria |
| fluoroquinolone antibiotic; monobactam; carbapenem; cephalosporin; glycylcycline; cephamycin; penam; tetracycline antibiotic; rifamycin antibiotic; phenicol antibiotic; triclosan; penem | antibiotic target alteration; antibiotic efflux; reduced permeability to antibiotic | Escherichia_coli_soxS | 1.16 × 10−06 | 1.33 × 10−07 | −3.12 | Acc | Acc | Bacteria |
| monobactam; carbapenem; cephalosporin; cephamycin; penam | antibiotic target alteration | Haemophilus_influenzae_PBP3 | 6.65 × 10−07 | 7.67 × 10−08 | −3.12 | Acc | Acc | Proteobacteria |
| peptide antibiotic | antibiotic efflux | YojI | 7.57 × 10−07 | 8.96 × 10−08 | −3.08 | Core | Acc | Proteobacteria |
| fluoroquinolone antibiotic; cephalosporin; glycylcycline; penam; tetracycline antibiotic; rifamycin antibiotic; phenicol antibiotic; triclosan | antibiotic efflux | acrB | 8.20 × 10−07 | 1.02 × 10−07 | −3.01 | Core | Acc | Proteobacteria; Enterobacteriaceae |
| macrolide antibiotic; fluoroquinolone antibiotic; cephalosporin; cephamycin; penam; tetracycline antibiotic | antibiotic efflux | H-NS | 1.02 × 10−06 | 1.27 × 10−07 | −3.00 | Acc | Acc | Proteobacteria |
| fluoroquinolone antibiotic | antibiotic efflux | mdtH | 7.05 × 10−07 | 8.90 × 10−08 | −2.99 | Acc | Acc | Proteobacteria |
| macrolide antibiotic; fluoroquinolone antibiotic; penam; tetracycline antibiotic | antibiotic efflux | evgS | 5.90 × 10−07 | 7.55 × 10−08 | −2.96 | Core | Acc | Proteobacteria; Enterobacteriaceae |
| phenicol antibiotic | antibiotic efflux | emrD | 9.01 × 10−07 | 1.19 × 10−07 | −2.92 | Core | Acc | Proteobacteria |
| aminocoumarin antibiotic | antibiotic efflux | mdtB | 9.06 × 10−07 | 1.21 × 10−07 | −2.90 | Core | Acc | Proteobacteria; Enterobacteriaceae |
| tetracycline antibiotic; benzalkonium chloride; rhodamine | antibiotic efflux | Escherichia_coli_mdfA | 5.75 × 10−07 | 8.33 × 10−08 | −2.79 | Core | Acc | Bacteria |
| macrolide antibiotic; fluoroquinolone antibiotic; penam | antibiotic efflux | mdtE | 9.49 × 10−07 | 1.39 × 10−07 | −2.77 | Acc | Acc | Proteobacteria |
| tetracycline antibiotic | antibiotic efflux | emrK | 5.73 × 10−07 | 9.31 × 10−08 | −2.62 | Acc | Acc | Proteobacteria |
| fosfomycin | antibiotic target alteration | Escherichia_coli_GlpT | 8.14 × 10−07 | 1.36 × 10−07 | −2.58 | Acc | Acc | Proteobacteria |
| cephalosporin; penam | antibiotic inactivation | Escherichia_coli_ampC | 9.61 × 10−07 | 1.61 × 10−07 | −2.58 | Acc | Acc | Proteobacteria/Enterobacteriaceae; Escherichia |
| peptide antibiotic | antibiotic target alteration | bacA | 7.34 × 10−07 | 1.26 × 10−07 | −2.54 | Acc | Acc | Proteobacteria |
| fosfomycin | antibiotic efflux | mdtG | 1.05 × 10−06 | 1.90 × 10−07 | −2.47 | Core | ND | Proteobacteria/Enterobacteriaceae; Escherichia |
| peptide antibiotic | antibiotic target alteration | PmrF | 1.13 × 10−06 | 2.07 × 10−07 | −2.44 | Core | Acc | Proteobacteria |
| fluoroquinolone antibiotic | antibiotic efflux | patA | 9.10 × 10−07 | 1.74 × 10−07 | −2.39 | Core | Acc | Proteobacteria/Firmicutes |
| nucleoside antibiotic; acridine dye | antibiotic efflux | mdtN | 8.21 × 10−07 | 1.59 × 10−07 | −2.37 | Core | Acc | Bacteria |
| aminocoumarin antibiotic | antibiotic efflux | mdtC | 1.32 × 10−06 | 2.84 × 10−07 | −2.22 | Core | Acc | Proteobacteria; Enterobacteriaceae |
| fluoroquinolone antibiotic; cephalosporin; cephamycin; penam | antibiotic efflux | AcrF | 1.63 × 10−06 | 3.82 × 10−07 | −2.10 | Core | Acc | Proteobacteria; Enterobacteriaceae |
| tetracycline antibiotic | antibiotic efflux | tetL | 3.40 × 10−06 | 8.65 × 10−07 | −1.98 | Core | Core | Bacteria |
| tetracycline antibiotic | antibiotic efflux | tetK | 7.68 × 10−06 | 2.25 × 10−06 | −1.77 | Core | Core | Bacteroidetes; Bacteroides |
| tetracycline antibiotic | antibiotic efflux | tet40 | 1.00 × 10−05 | 3.71 × 10−06 | −1.43 | Core | Core | Firmicutes; Clostridiales |
| tetracycline antibiotic | antibiotic target protection | tetBP | 1.70 × 10−06 | 6.73 × 10−07 | −1.34 | Core | Core | Bacteria |
| tetracycline antibiotic | antibiotic target protection | tet44 | 8.11 × 10−05 | 3.34 × 10−05 | −1.28 | Core | Core | Bacteroidetes; Bacteroides |
| fluoroquinolone antibiotic | antibiotic efflux | pmrA | 1.02 × 10−05 | 4.33 × 10−06 | −1.24 | Core | Core | Bacteroidetes; Bacteroides |
| tetracycline antibiotic | antibiotic target protection | tetO | 1.88 × 10−05 | 8.25 × 10−06 | −1.19 | Core | Core | Bacteria |
| tetracycline antibiotic | antibiotic target protection | tetW/N/W | 2.26 × 10−04 | 1.57 × 10−04 | −0.52 | Core | Core | Bacteria |
| cephalosporin; penam | antibiotic inactivation | OXA-7 | 4.71 × 10−07 | 1.03 × 10−06 | 1.13 | Acc | Core | Bacteroidetes; Bacteroides |
| macrolide antibiotic; lincosamide antibiotic | antibiotic target alteration | RlmAII | 1.77 × 10−07 | 7.68 × 10−07 | 2.12 | Acc | Core | Bacteroidetes; Prevotella bryantii |
| monobactam; carbapenem; cephalosporin; cephamycin; penam; penem | antibiotic inactivation | GIM-2 | 3.25 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Unknown |
| cephalosporin; penam | antibiotic inactivation | OXA-136 | 7.55 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Spirochaetes; Brachyspira pilosicoli |
| cephalosporin; penam | antibiotic inactivation | OXA-244 | 1.58 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Unknown |
| monobactam; cephalosporin; penam; penem | antibiotic inactivation | TEM-127 | 1.48 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Bacteria |
| monobactam; cephalosporin; penam; penem | antibiotic inactivation | TEM-219 | 1.39 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Enterobacteriaceae |
| streptogramin antibiotic; pleuromutilin antibiotic | antibiotic efflux | vgaC | 8.45 × 10−07 | 0.00 × 10+00 | Depleted | Acc | ND | Proteobacteria; Enterobacteriaceae |
| aminoglycoside antibiotic | antibiotic inactivation | AAC6-Ii | 0.00 × 10+00 | 3.06 × 10−07 | Detected | Acc | Acc | Unknown |
| carbapenem; cephalosporin; cephamycin; penam | antibiotic inactivation | ACT-30 | 0.00 × 10+00 | 4.95 × 10−07 | Detected | ND | Acc | Unknown |
| aminoglycoside antibiotic | antibiotic inactivation | APH3-IVa | 0.00 × 10+00 | 6.40 × 10−08 | Detected | ND | Acc | Unknown |
| penam | antibiotic inactivation | CARB-3 | 0.00 × 10+00 | 1.55 × 10−07 | Detected | ND | Acc | Unknown |
| tetracycline antibiotic | antibiotic efflux | tcr3 | 0.00 × 10+00 | 1.33 × 10−07 | Detected | ND | Acc | Ruminococcaceae |
| streptogramin antibiotic | antibiotic inactivation | vgbC | 0.00 × 10+00 | 2.40 × 10−07 | Detected | ND | Acc | Ruminococcaceae |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corrigan, A.; McCooey, P.; Taylor-Pickard, J.; Stockdale, S.; Murphy, R. Breaking the Cycle: A Yeast Mannan-Rich Fraction Beneficially Modulates Egg Quality and the Antimicrobial Resistome Associated with Layer Hen Caecal Microbiomes under Commercial Conditions. Microorganisms 2024, 12, 1562. https://doi.org/10.3390/microorganisms12081562
Corrigan A, McCooey P, Taylor-Pickard J, Stockdale S, Murphy R. Breaking the Cycle: A Yeast Mannan-Rich Fraction Beneficially Modulates Egg Quality and the Antimicrobial Resistome Associated with Layer Hen Caecal Microbiomes under Commercial Conditions. Microorganisms. 2024; 12(8):1562. https://doi.org/10.3390/microorganisms12081562
Chicago/Turabian StyleCorrigan, Aoife, Paula McCooey, Jules Taylor-Pickard, Stephen Stockdale, and Richard Murphy. 2024. "Breaking the Cycle: A Yeast Mannan-Rich Fraction Beneficially Modulates Egg Quality and the Antimicrobial Resistome Associated with Layer Hen Caecal Microbiomes under Commercial Conditions" Microorganisms 12, no. 8: 1562. https://doi.org/10.3390/microorganisms12081562