
Genetic Diversity in Salmonella enterica in Outbreaks of Foodborne and Zoonotic Origin in the USA in 2006–2017

 ,
, 
 , , , and
, , , and
Abstract
1. Introduction
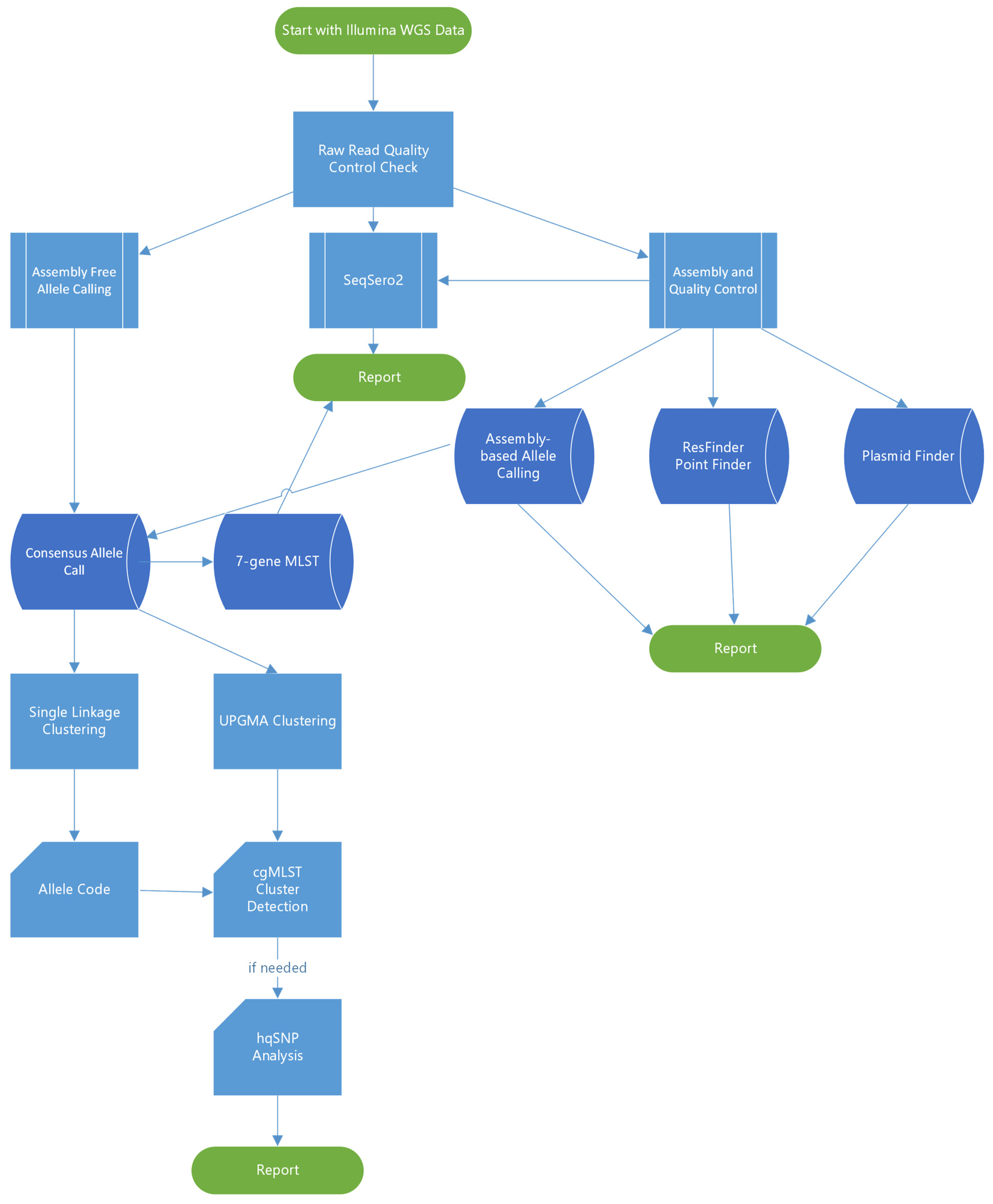
2. Materials and Methods
2.1. Selection of Outbreaks and Bacterial Strains
2.2. Whole Genome Sequencing (WGS)
2.3. Data Analysis
3. Results
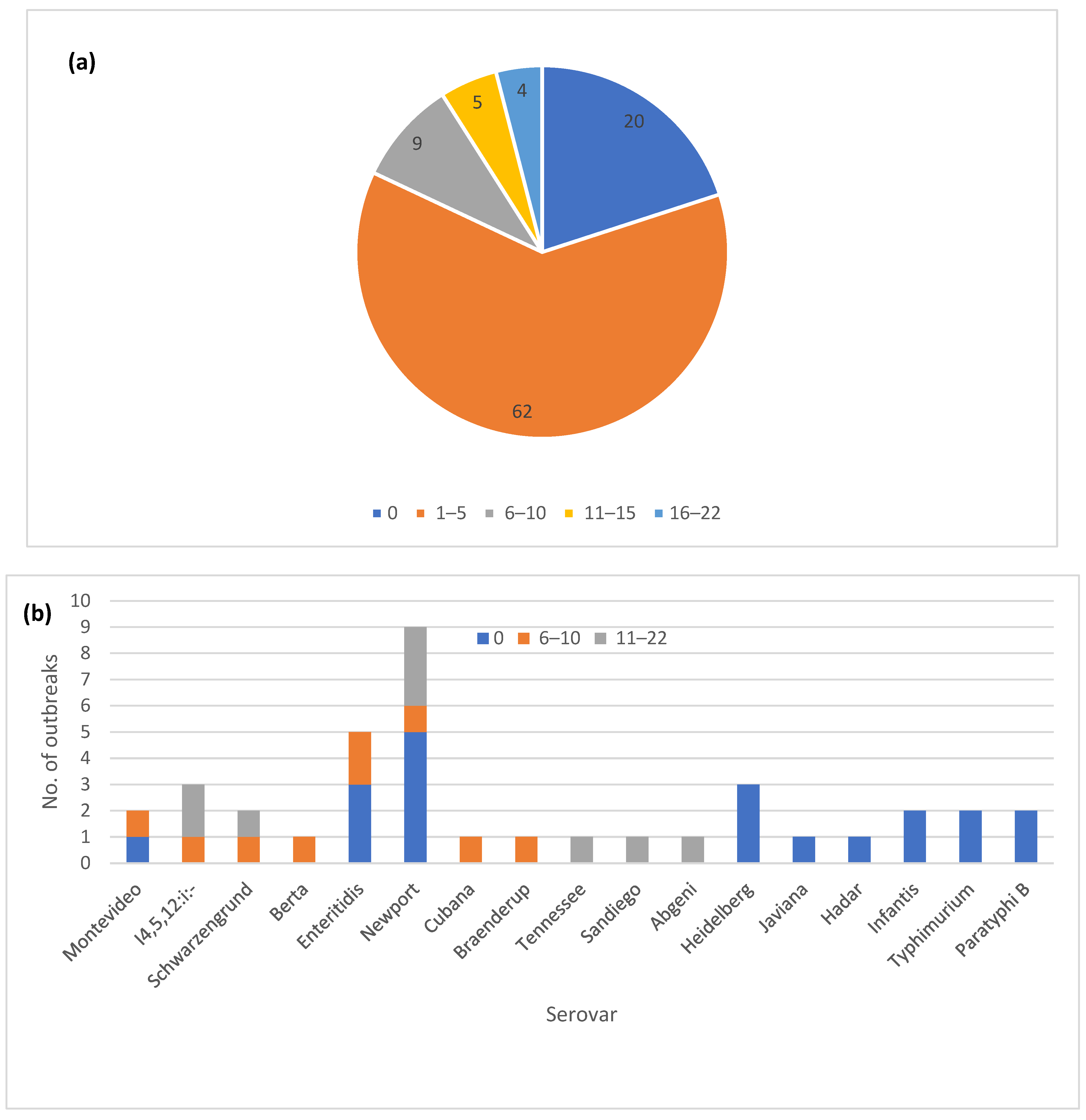
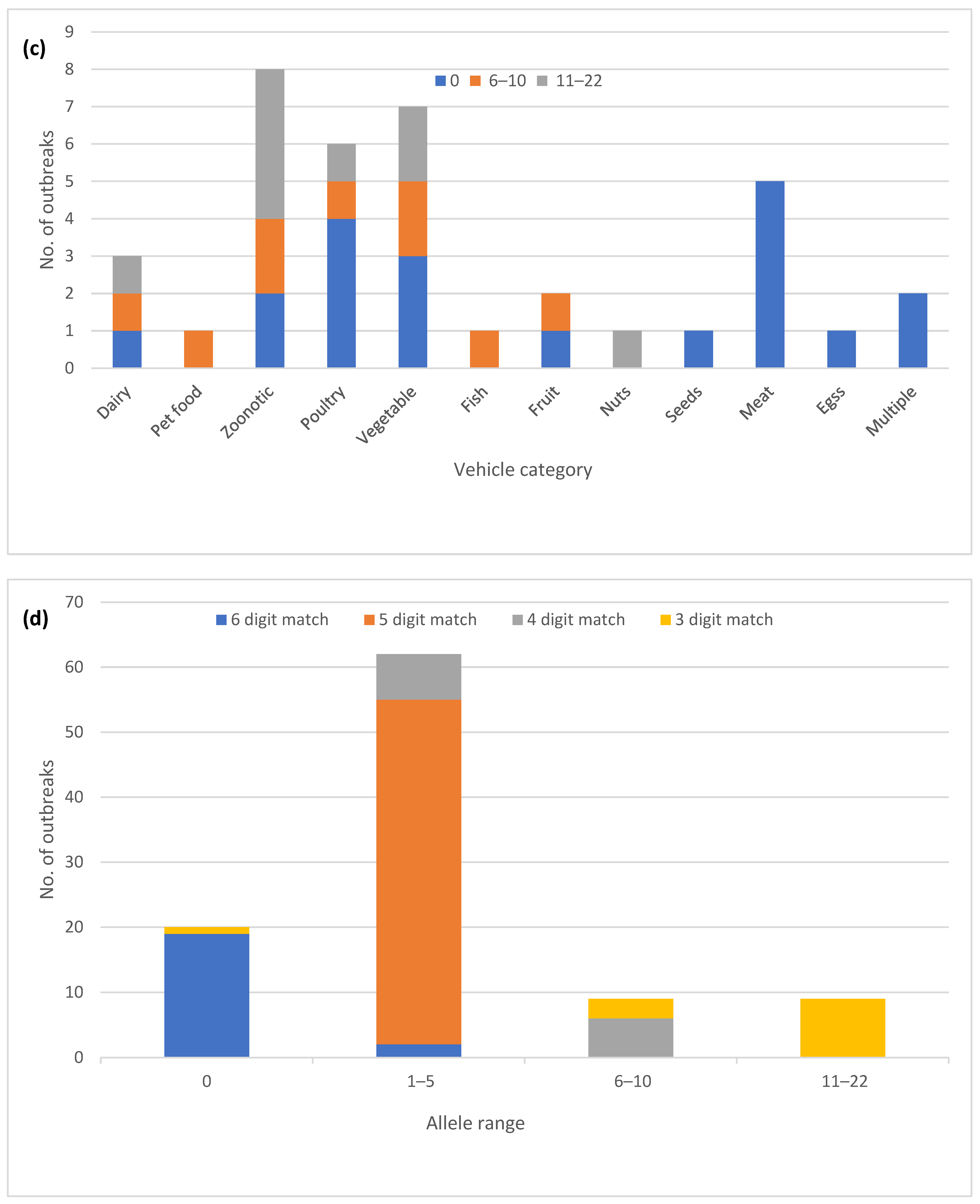
3.1. General Description of the Outbreaks
3.2. Diversity of cgMLST Profiles
3.3. Confirmatory hqSNP Analysis
3.4. Correlation of cgMLST and PFGE
3.5. Diversity of 7-Gene MLST
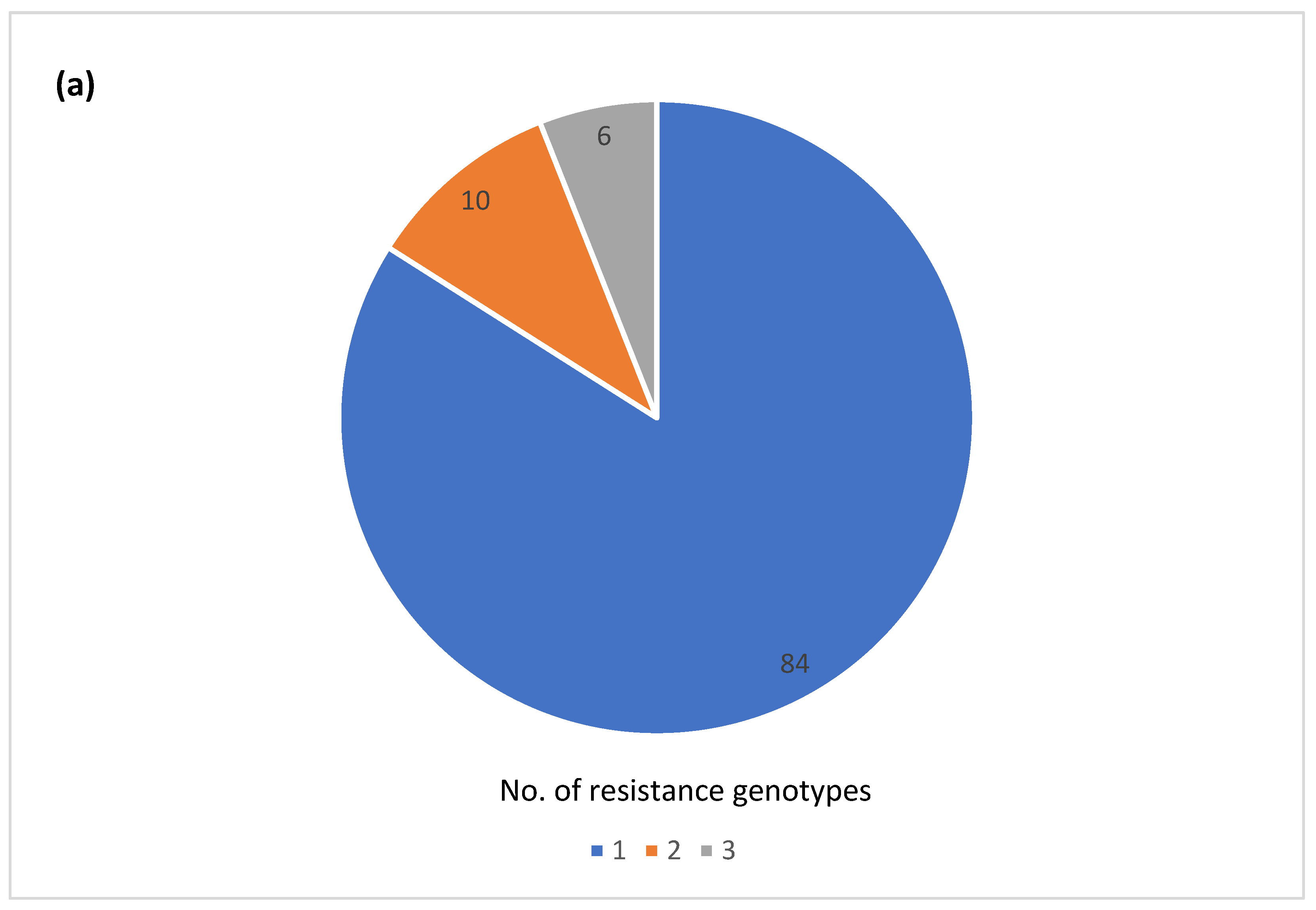
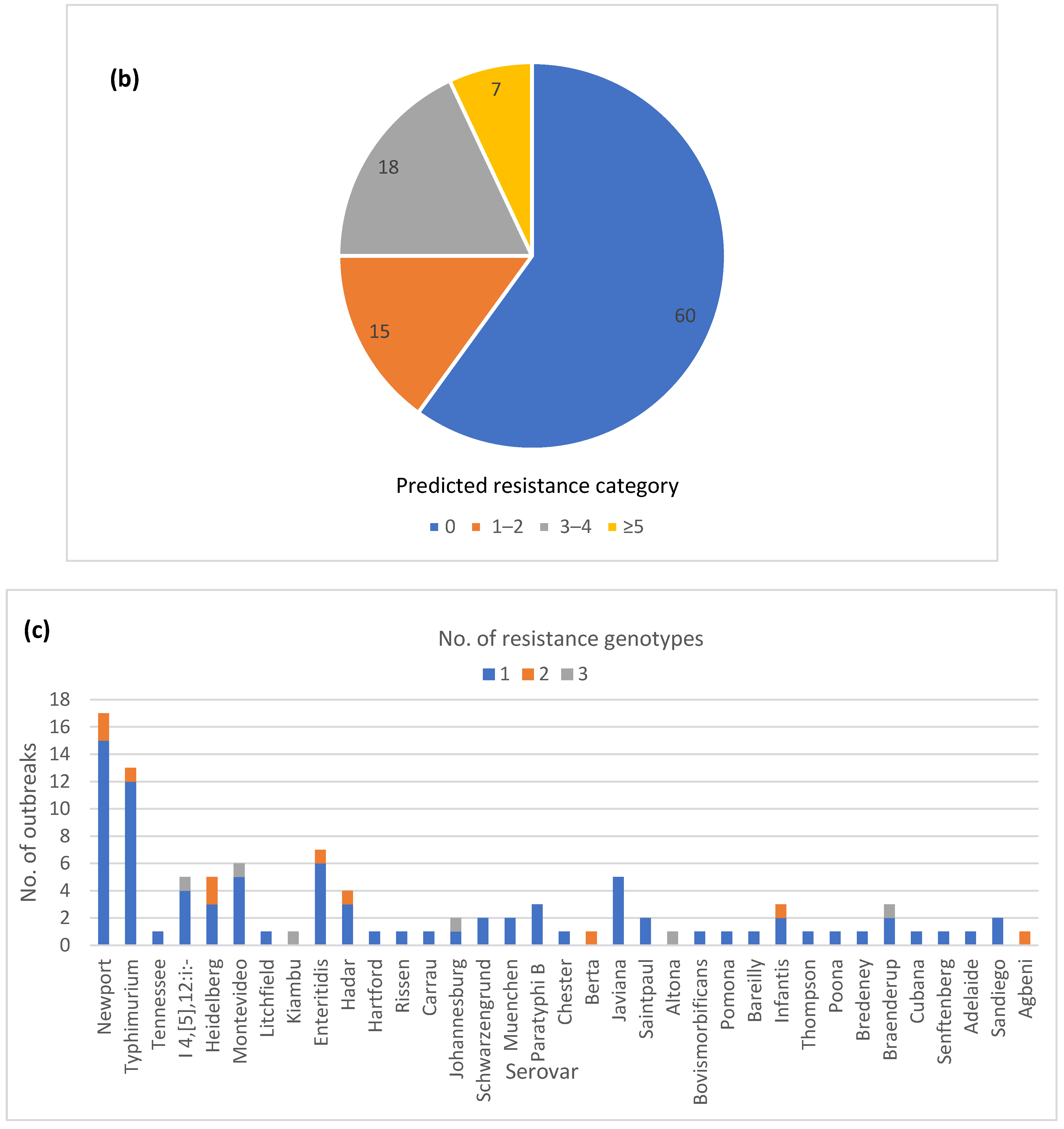
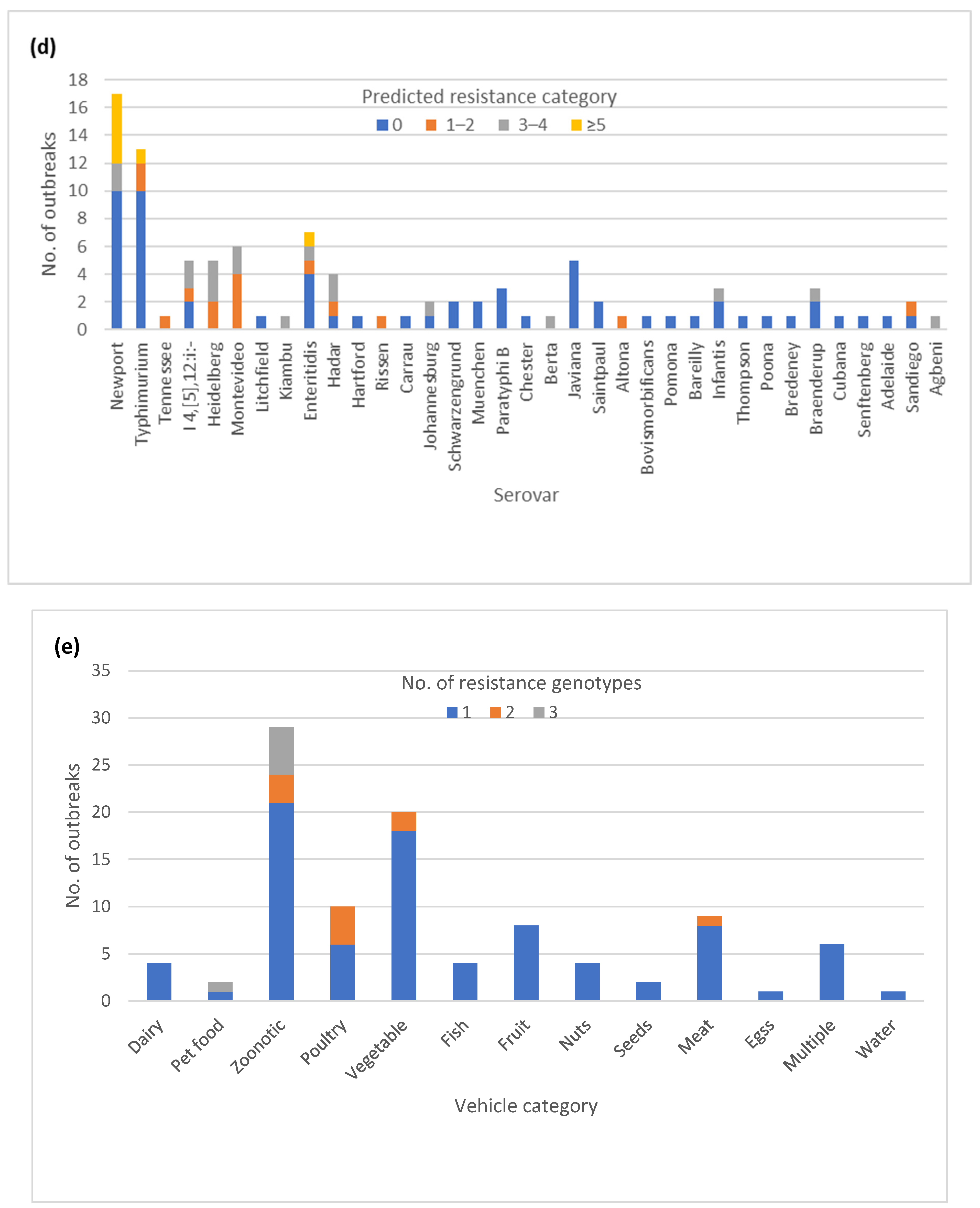
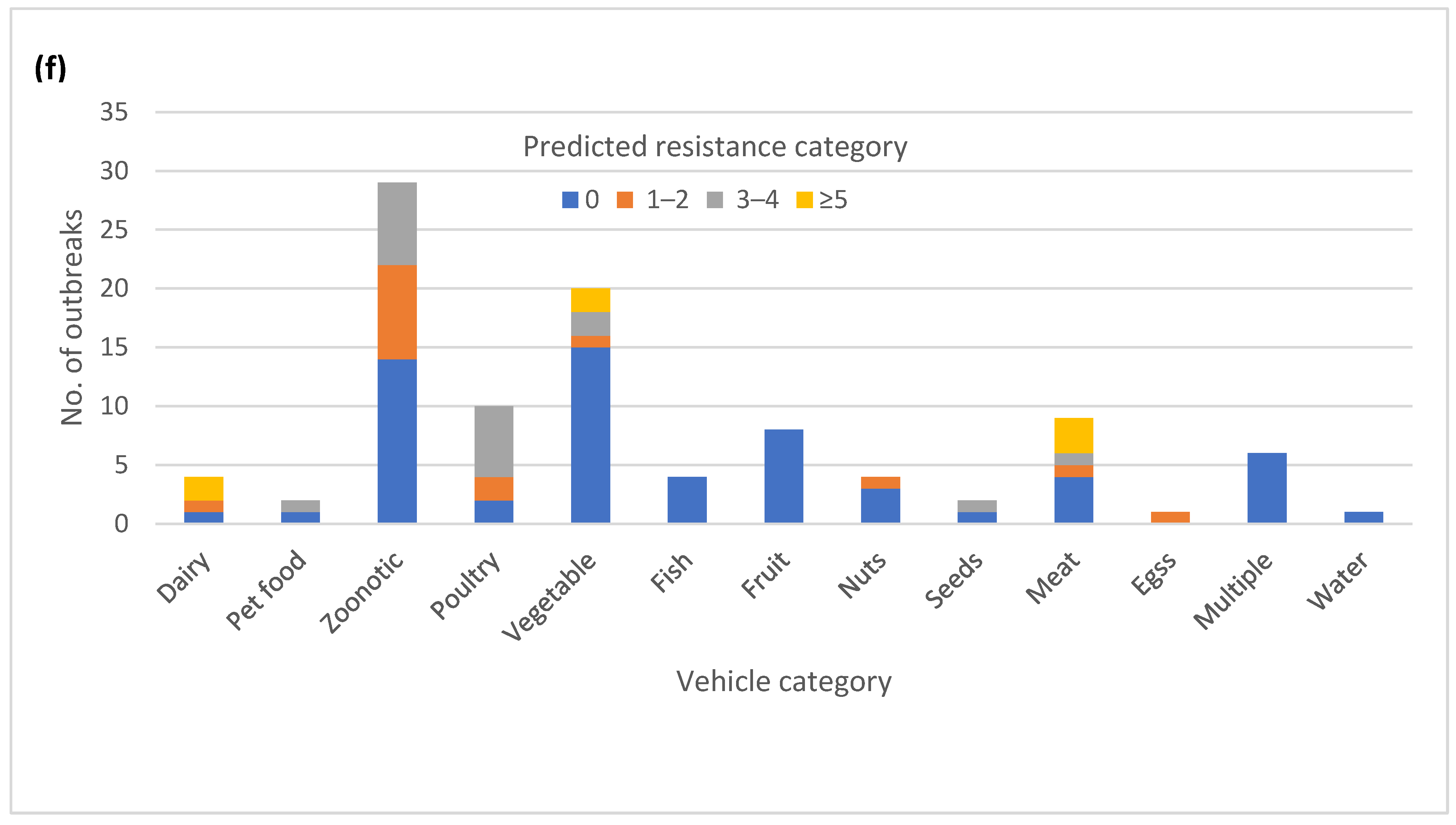
3.6. Diversity of AR
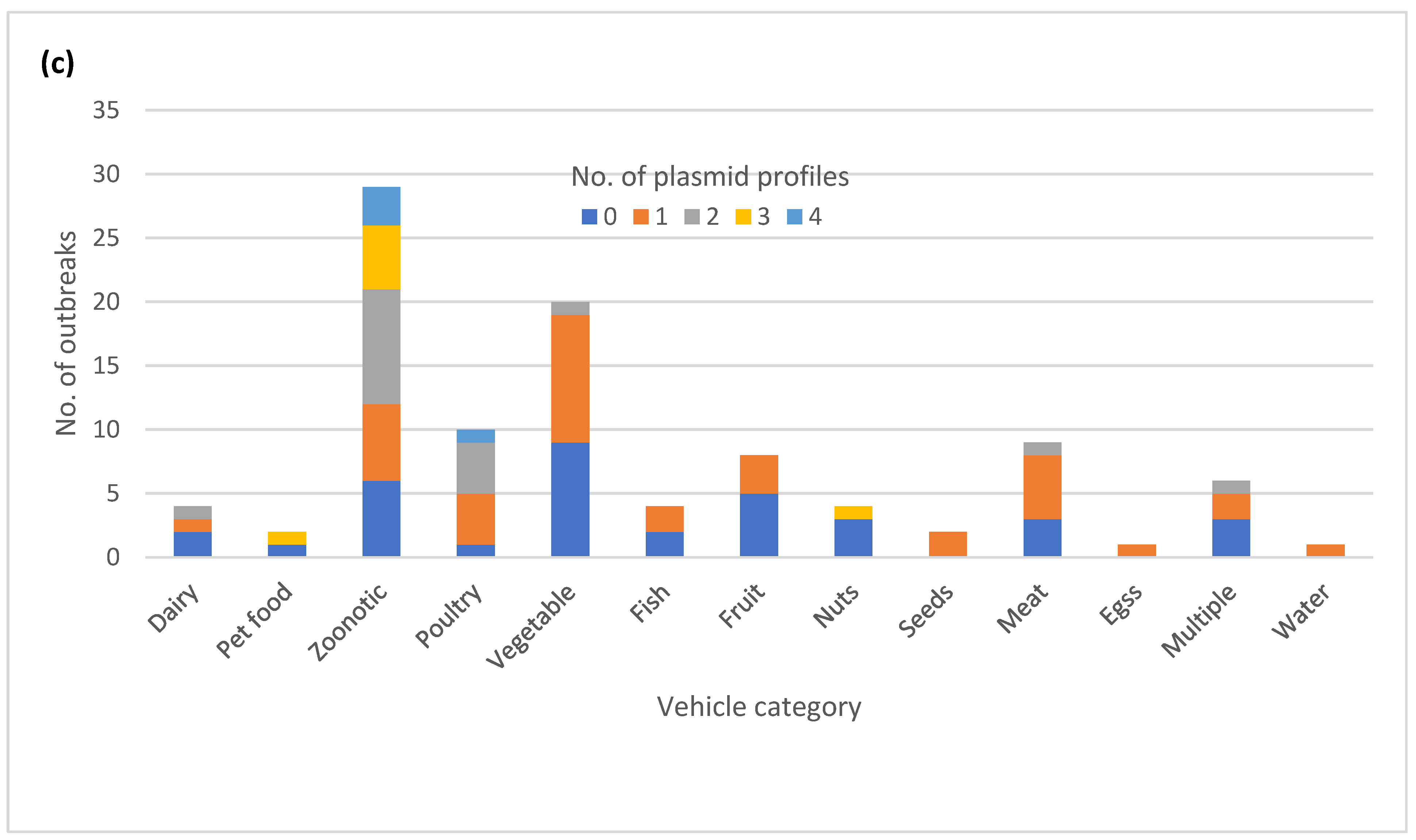
3.7. Diversity of Plasmid Profiles
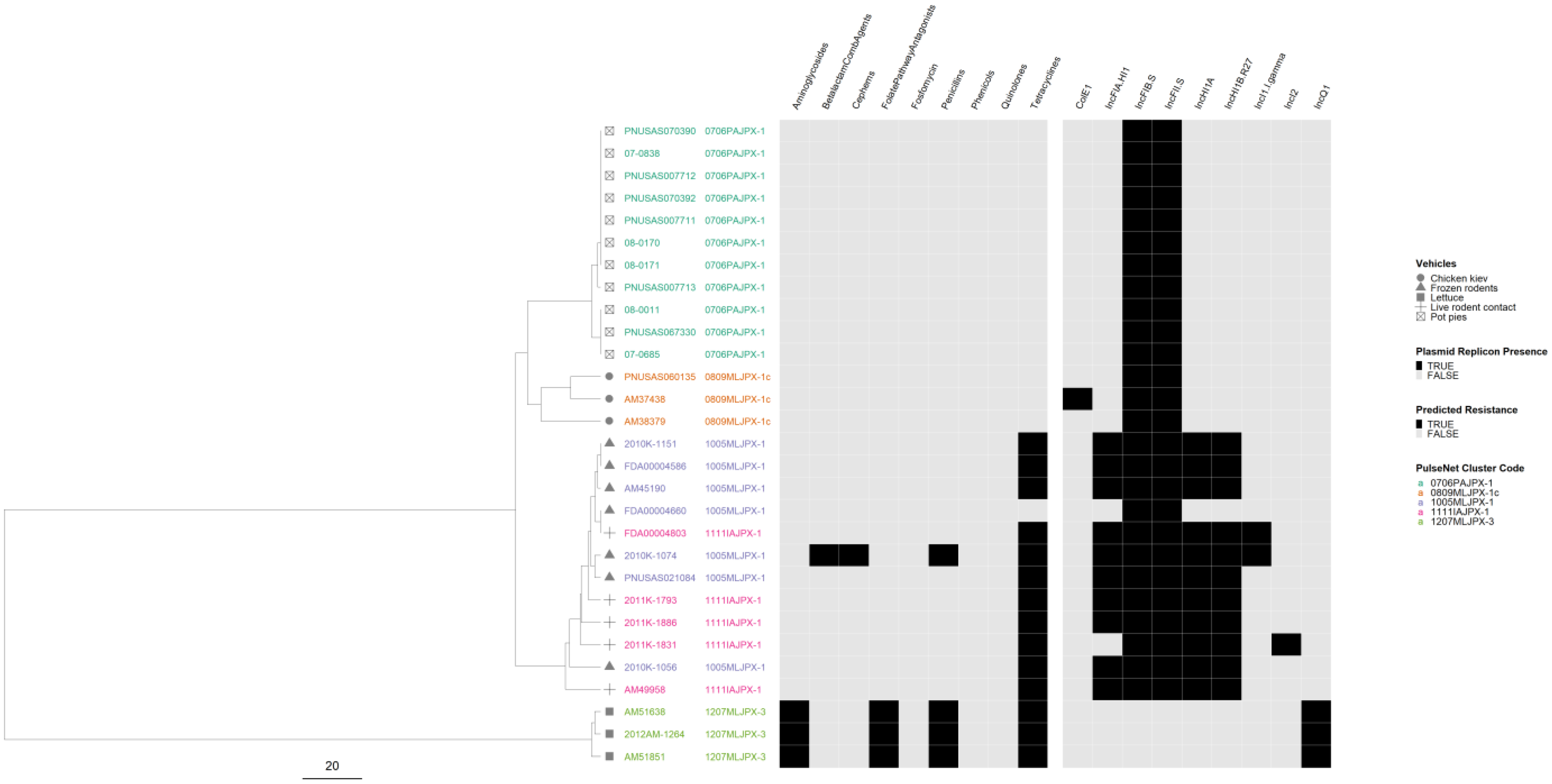
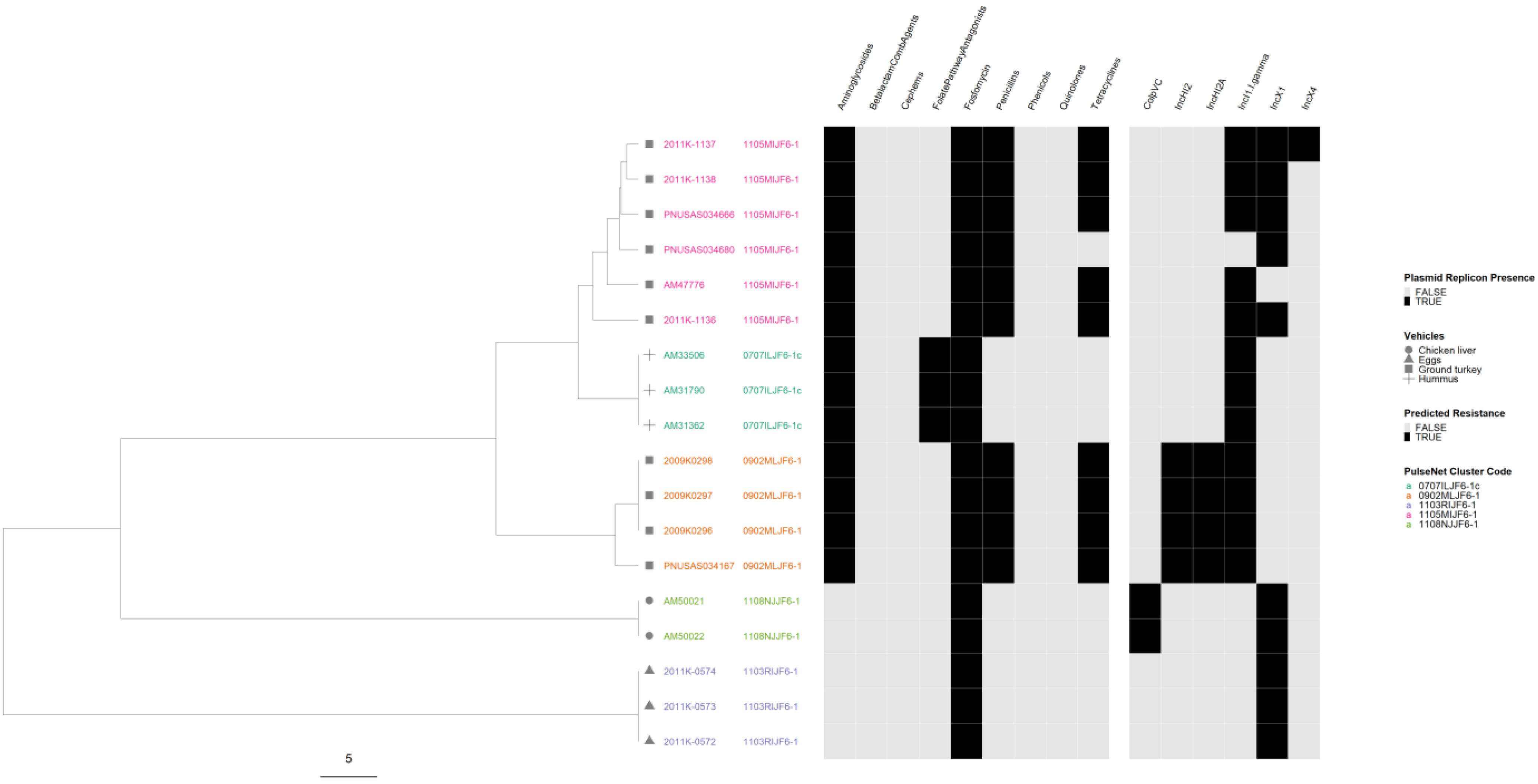
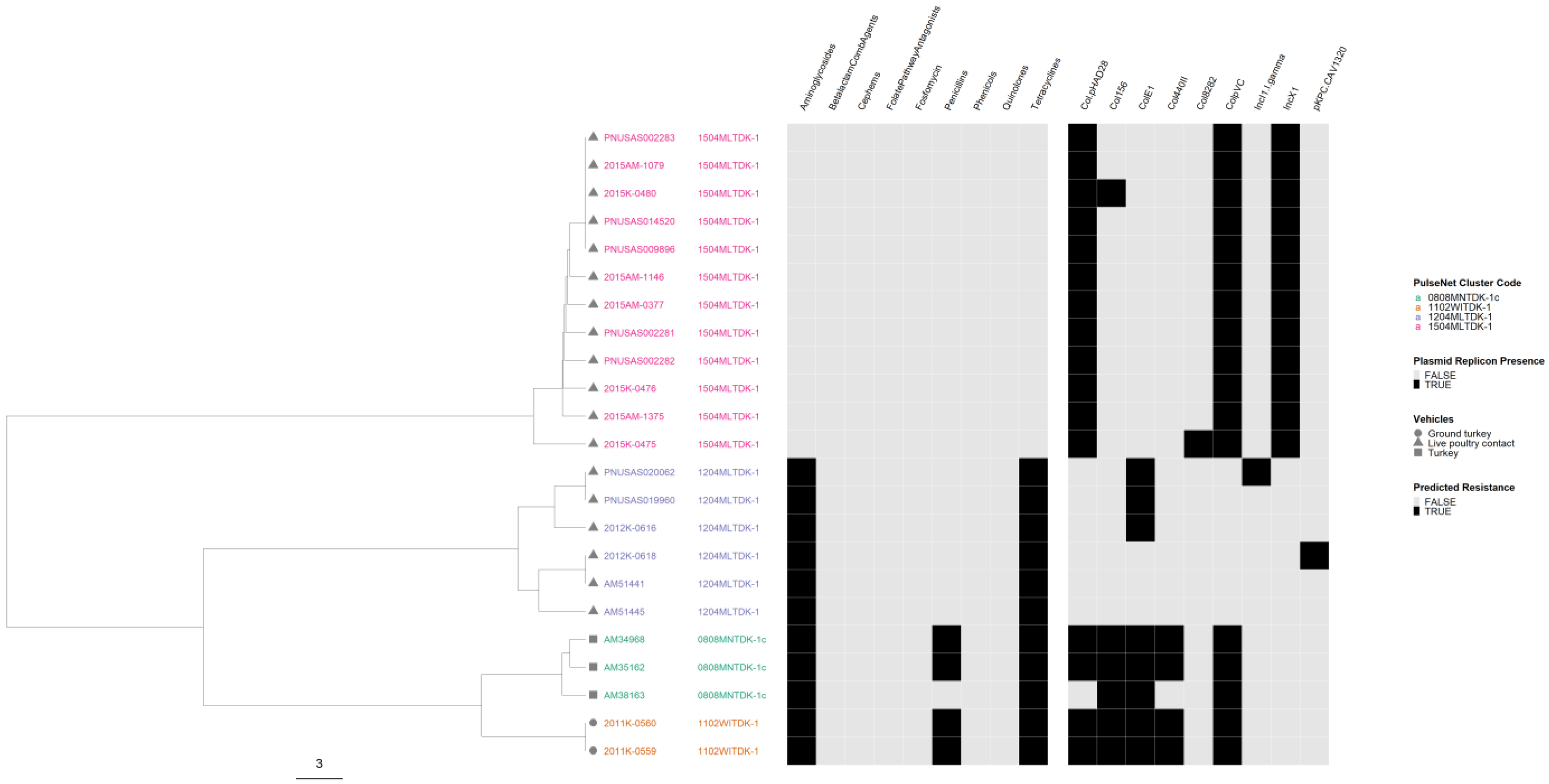
3.8. Correlation of cgMLST Clustering with AR and Plasmid Profiles for Selected Serovars
4. Discussion
4.1. Diversity of cgMLST Profiles and Confirmatory hqSNP Analysis
4.2. Correlation of cgMLST with Legacy Subtyping Methods
4.3. Diversity of AR and Plasmid Profiles
4.4. Correlation of cgMLST Clustering with AR and Plasmid Profiles for Selected Serovars
4.5. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne Illness Acquired in the United States—Major Pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Salmonella Surveillance Overview. 2011. Available online: https://www.cdc.gov/nationalsurveillance/salmonella-surveillance.html (accessed on 14 March 2024).
- Baker, D.J.; Robbins, A.; Newman, J.; Anand, M.; Wolfgang, W.J.; Mendez-Vallellanes, D.V.; Wirth, S.E.; Mingle, L.A. Challenges Associated with Investigating Salmonella Enteritidis with Low Genomic Diversity in New York State: The Impact of Adjusting Analytical Methods and Correlation with Epidemiological Data. Foodborne Pathog. Dis. 2023, 20, 230–236. [Google Scholar] [CrossRef]
- Timme, R.E.; Pettengill, J.B.; Allard, M.W.; Strain, E.; Barrangou, R.; Wehnes, C.; Van Kessel, J.S.; Karns, J.S.; Musser, S.M.; Brown, E.W. Phylogenetic Diversity of the Enteric Pathogen Salmonella enterica Subsp. enterica Inferred from Genome-Wide Reference-Free SNP Characters. Genome Biol. Evol. 2013, 5, 2109–2123. [Google Scholar] [CrossRef] [PubMed]
- Tolar, B.; Joseph, L.A.; Schroeder, M.N.; Stroika, S.; Ribot, E.M.; Hise, K.B.; Gerner-Smidt, P. An Overview of PulseNet USA Databases. Foodborne Pathog. Dis. 2019, 16, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, E.; Date, K.; Medus, C.; Meyer, S.; Miller, B.; Kim, C.; Nowicki, S.; Cosgrove, S.; Sweat, D.; Phan, Q.; et al. Salmonella Typhimurium Infections Associated with Peanut Products. N. Engl. J. Med. 2011, 365, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Slayton, R.B.; Turabelidze, G.; Bennett, S.D.; Schwensohn, C.A.; Yaffee, A.Q.; Khan, F.; Butler, C.; Trees, E.; Ayers, T.L.; Davis, M.L.; et al. Outbreak of Shiga Toxin-Producing Escherichia coli (STEC) O157:H7 Associated with Romaine Lettuce Consumption, 2011. PLoS ONE 2013, 8, e55300. [Google Scholar] [CrossRef] [PubMed]
- Besser, J.M.; Carleton, H.A.; Trees, E.; Stroika, S.G.; Hise, K.; Wise, M.; Gerner-Smidt, P. Interpretation of Whole-Genome Sequencing for Enteric Disease Surveillance and Outbreak Investigation. Foodborne Pathog. Dis. 2019, 16, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Ribot, E.M.; Fair, M.A.; Gautom, R.; Cameron, D.N.; Hunter, S.B.; Swaminathan, B.; Barrett, T.J. Standardization of Pulsed-Field Gel Electrophoresis Protocols for the Subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog. Dis. 2006, 3, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.C.; Bazaco, M.C.; Parker, C.C.; Dewey-Mattia, D.; Golden, N.; Jones, K.; Klontz, K.; Travis, C.; Kufel, J.Z.; Cole, D. An Updated Scheme for Categorizing Foods Implicated in Foodborne Disease Outbreaks: A Tri-Agency Collaboration. Foodborne Pathog. Dis. 2017, 14, 701–710. [Google Scholar] [CrossRef]
- PulseNet. Laboratory Standard Operating Procedure for PulseNet Total DNA Extraction and Quality Control of Purified DNA Extracts. 2021. Available online: https://www.aphl.org/programs/food_safety/Pages/PulseNet-International-SOPs.aspx (accessed on 1 November 2023).
- PulseNet. Laboratory Standard Operating Procedure for PulseNet Nextera XT Library Preparation. 2021. Available online: https://www.aphl.org/programs/food_safety/Pages/PulseNet-International-SOPs.aspx (accessed on 1 November 2023).
- PulseNet. PulseNet Standard Operating Procedure for DNA Library Preparation Using the Illumina® DNA Prep Kit. 2023. Available online: https://www.aphl.org/programs/food_safety/Pages/PulseNet-International-SOPs.aspx (accessed on 1 November 2023).
- PulseNet. Laboratory Standard Operating Procedure for Whole Genome Sequencing on MiSeq. 2023. Available online: https://www.aphl.org/programs/food_safety/Pages/PulseNet-International-SOPs.aspx (accessed on 1 November 2023).
- Zhou, Z.; Alikhan, N.-F.; Mohamed, K.; Fan, Y.; The Agama Study Group; Achtman, M.; Brown, D.; Chattaway, M.; Dallman, T.; Delahay, R.; et al. The EnteroBase User’s Guide, with Case Studies on Salmonella Transmissions, Yersinia pestis Phylogeny, and Escherichia Core Genomic Diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef]
- Leeper, M.M.; Tolar, B.M.; Griswold, T.; Vidyaprakash, E.; Hise, K.B.; Williams, G.M.; Im, S.B.; Chen, J.C.; Pouseele, H.; Carleton, H.A. Evaluation of Whole and Core Genome Multilocus Sequence Typing Allele Schemes for Salmonella enterica Outbreak Detection in a National Surveillance Network, PulseNet USA. Front. Microbiol. 2023, 14, 1254777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Petkau, A.; Avery, B.P.; Chen, J.C.; Folster, J.P.; Carson, C.A.; Kearney, A.; Nadon, C.; Mabon, P.; Thiessen, J.; et al. Correlation between Phenotypic and In Silico Detection of Antimicrobial Resistance in Salmonella enterica in Canada Using Staramr. Microorganisms 2022, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico PMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). In Horizontal Gene Transfer; De La Cruz, F., Ed.; Springer: New York, NY, USA, 2020; Volume 2075, pp. 285–294. ISBN 9781493998760. [Google Scholar]
- Katz, L.S.; Griswold, T.; Williams-Newkirk, A.J.; Wagner, D.; Petkau, A.; Sieffert, C.; Van Domselaar, G.; Deng, X.; Carleton, H.A. A Comparative Analysis of the Lyve-SET Phylogenomics Pipeline for Genomic Epidemiology of Foodborne Pathogens. Front. Microbiol. 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pettengill, J.B.; Pightling, A.; Timme, R.; Allard, M.; Strain, E.; Rand, H. Genetic Diversity of Salmonella and Listeria Isolates from Food Facilities. J. Food Prot. 2018, 81, 2082–2089. [Google Scholar] [CrossRef]
- Dallman, T.; Ashton, P.; Schafer, U.; Jironkin, A.; Painset, A.; Shaaban, S.; Hartman, H.; Myers, R.; Underwood, A.; Jenkins, C.; et al. SnapperDB: A Database Solution for Routine Sequencing Analysis of Bacterial Isolates. Bioinformatics 2018, 34, 3028–3029. [Google Scholar] [CrossRef] [PubMed]
- Pightling, A.W.; Rand, H.; Pettengill, J. Using Evolutionary Analyses to Refine Whole-Genome Sequence Match Criteria. Front. Microbiol. 2022, 13, 797997. [Google Scholar] [CrossRef] [PubMed]
- Gerner-Smidt, P.; Besser, J.; Concepción-Acevedo, J.; Folster, J.P.; Huffman, J.; Joseph, L.A.; Kucerova, Z.; Nichols, M.C.; Schwensohn, C.A.; Tolar, B. Whole Genome Sequencing: Bridging One-Health Surveillance of Foodborne Diseases. Front. Public Health 2019, 7, 172. [Google Scholar]
- Nichols, M.; Stevenson, L.; Koski, L.; Basler, C.; Wise, M.; Whitlock, L.; Francois Watkins, L.; Friedman, C.R.; Chen, J.; Tagg, K.; et al. Detecting National Human Enteric Disease Outbreaks Linked to Animal Contact in the United States of America. Rev. Sci. Tech. OIE 2020, 39, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.C.; Nguyen, T.-A.; Adams, J.K.; Garrett, N.M.; Bopp, C.A.; Baker, J.B.; McNeil, C.; Torres, P.; Ettestad, P.J.; Erdman, M.M.; et al. Multistate Outbreak of Human Salmonella Typhimurium Infections Linked to Live Poultry from Agricultural Feed Stores and Mail-Order Hatcheries, United States 2013. One Health 2016, 2, 144–149. [Google Scholar] [CrossRef]
- Basler, C.; Nguyen, T.-A.; Anderson, T.C.; Hancock, T.; Behravesh, C.B. Outbreaks of Human Salmonella Infections Associated with Live Poultry, United States, 1990–2014. Emerg. Infect. Dis. 2016, 22, 1705–1711. [Google Scholar] [CrossRef]
- Robertson, S.A.; Sidge, J.L.; Koski, L.; Hardy, M.C.; Stevenson, L.; Signs, K.; Stobierski, M.G.; Bidol, S.; Donovan, D.; Soehnlen, M.; et al. Onsite Investigation at a Mail-Order Hatchery Following a Multistate Salmonella Illness Outbreak Linked to Live Poultry—United States, 2018. Poult. Sci. 2019, 98, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, G.S.; Habrun, C.; Nemechek, K.; Gollarza, L.; Ellison, Z.; Tolar, B.; Koski, L.; Brandenburg, J.M.; Salah, Z.; Palacios, A.; et al. Multistate Outbreaks of Salmonellosis Linked to Contact with Backyard Poultry—United States, 2015–2022. Zoonoses Public Health 2024, 1–15. [Google Scholar] [CrossRef]
- Cartwright, E.J.; Nguyen, T.; Melluso, C.; Ayers, T.; Lane, C.; Hodges, A.; Li, X.; Quammen, J.; Yendell, S.J.; Adams, J.; et al. A Multistate Investigation of Antibiotic-Resistant Salmonella Enterica Serotype I 4,[5],12:I:- Infections as Part of an International Outbreak Associated with Frozen Feeder Rodents. Zoonoses Public Health 2016, 63, 62–71. [Google Scholar] [CrossRef]
- Barrett, T.J.; Gerner-Smidt, P.; Swaminathan, B. Interpretation of Pulsed-Field Gel Electrophoresis Patterns in Foodborne Disease Investigations and Surveillance. Foodborne Pathog. Dis. 2006, 3, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Stevens, E.L.; Carleton, H.A.; Beal, J.; Tillman, G.E.; Lindsey, R.L.; Lauer, A.C.; Pightling, A.; Jarvis, K.G.; Ottesen, A.; Ramachandran, P.; et al. Use of Whole Genome Sequencing by the Federal Interagency Collaboration for Genomics for Food and Feed Safety in the United States. J. Food Prot. 2022, 85, 755–772. [Google Scholar] [CrossRef]
- Glenn, L.M.; Lindsey, R.L.; Folster, J.P.; Pecic, G.; Boerlin, P.; Gilmour, M.W.; Harbottle, H.; Zhao, S.; McDermott, P.F.; Fedorka-Cray, P.J.; et al. Antimicrobial Resistance Genes in Multidrug-Resistant Salmonella Enterica Isolated from Animals, Retail Meats, and Humans in the United States and Canada. Microb. Drug Resist. 2013, 19, 175–184. [Google Scholar] [CrossRef]
- Kaldhone, P.R.; Carlton, A.; Aljahdali, N.; Khajanchi, B.K.; Sanad, Y.M.; Han, J.; Deck, J.; Ricke, S.C.; Foley, S.L. Evaluation of Incompatibility Group I1 (IncI1) Plasmid-Containing Salmonella Enterica and Assessment of the Plasmids in Bacteriocin Production and Biofilm Development. Front. Vet. Sci. 2019, 6, 298. [Google Scholar] [CrossRef]
- Canning, M.; Birhane, M.G.; Dewey-Mattia, D.; Lawinger, H.; Cote, A.; Gieraltowski, L.; Schwensohn, C.; Tagg, K.A.; Francois Watkins, L.K.; Park Robyn, M.; et al. Salmonella Outbreaks Linked to Beef, United States, 2012–2019. J. Food Prot. 2023, 86, 100071. [Google Scholar] [CrossRef]
- Hull, D.M.; Harrell, E.; Harden, L.; Thakur, S. Multidrug Resistance and Virulence Genes Carried by Mobile Genomic Elements in Salmonella enterica Isolated from Live Food Animals, Processed, and Retail Meat in North Carolina, 2018–2019. Int. J. Food Microbiol. 2022, 378, 109821. [Google Scholar] [CrossRef]
- Nichols, M.; Gollarza, L.; Sockett, D.; Aulik, N.; Patton, E.; Francois Watkins, L.K.; Gambino-Shirley, K.J.; Folster, J.P.; Chen, J.C.; Tagg, K.A.; et al. Outbreak of Multidrug-Resistant Salmonella Heidelberg Infections Linked to Dairy Calf Exposure, United States, 2015–2018. Foodborne Pathog. Dis. 2022, 19, 199–208. [Google Scholar] [CrossRef]
- Phillips, C.; Chapman, B.; Agunos, A.; Carson, C.A.; Parmley, E.J.; Reid-Smith, R.J.; Smith, B.A.; Murphy, C.P. A Scoping Review of Factors Potentially Linked with Antimicrobial-Resistant Bacteria from Turkeys (IAM.AMR Project). Epidemiol. Infect. 2022, 150, e153. [Google Scholar] [CrossRef]
- Varga, C.; Guerin, M.T.; Brash, M.L.; Slavic, D.; Boerlin, P.; Susta, L. Antimicrobial Resistance in Fecal Escherichia coli and Salmonella enterica Isolates: A Two-Year Prospective Study of Small Poultry Flocks in Ontario, Canada. BMC Vet. Res. 2019, 15, 464. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC); European Food Safety Authority (EFSA); European Medicines Agency (EMA). Antimicrobial Consumption and Resistance in Bacteria from Humans and Food—Producing Animals (JIACRA IV—2019−2021) [WWW Document]. 2024. Available online: https://www.ecdc.europa.eu/en/publications-data/antimicrobial-consumption-and-resistance-bacteria-humans-and-food-producing (accessed on 14 March 2024).
- Waltenburg, M.A.; Perez, A.; Salah, Z.; Karp, B.E.; Whichard, J.; Tolar, B.; Gollarza, L.; Koski, L.; Blackstock, A.; Basler, C.; et al. Multistate Reptile- and amphibian-associated Salmonellosis Outbreaks in Humans, United States, 2009–2018. Zoonoses Public Health 2022, 69, 925–937. [Google Scholar] [CrossRef]
- Pitout, J.D.D.; Reisbig, M.D.; Mulvey, M.; Chui, L.; Louie, M.; Crowe, L.; Church, D.L.; Elsayed, S.; Gregson, D.; Ahmed, R.; et al. Association between Handling of Pet Treats and Infection with Salmonella enterica Serotype Newport Expressing the AmpC β-Lactamase, CMY-2. J. Clin. Microbiol. 2003, 41, 4578–4582. [Google Scholar] [CrossRef]
- Turcotte, M.R.; Smith, J.T.; Li, J.; Zhang, X.; Wolfe, K.L.; Gao, F.; Benton, C.S.; Andam, C.P. Genome Characteristics of Clinical Salmonella enterica Population from a State Public Health Laboratory, New Hampshire, USA, 2017–2020. BMC Genom. 2022, 23, 537. [Google Scholar] [CrossRef]
- Lee, W.W.Y.; Mattock, J.; Greig, D.R.; Langridge, G.C.; Baker, D.; Bloomfield, S.; Mather, A.E.; Wain, J.R.; Edwards, A.M.; Hartman, H.; et al. Characterization of a PESI-like Plasmid and Analysis of Multidrug-Resistant Salmonella enterica Infantis Isolates in England and Wales. Microb. Genom. 2021, 7, 000658. [Google Scholar] [CrossRef]
- Peter, S.; Bosio, M.; Gross, C.; Bezdan, D.; Gutierrez, J.; Oberhettinger, P.; Liese, J.; Vogel, W.; Dörfel, D.; Berger, L.; et al. Tracking of Antibiotic Resistance Transfer and Rapid Plasmid Evolution in a Hospital Setting by Nanopore Sequencing. mSphere 2020, 5, e00525-20. [Google Scholar] [CrossRef]
- Liu, C.M.; Aziz, M.; Park, D.E.; Wu, Z.; Stegger, M.; Li, M.; Wang, Y.; Schmidlin, K.; Johnson, T.J.; Koch, B.J.; et al. Using Source-Associated Mobile Genetic Elements to Identify Zoonotic Extraintestinal E. coli Infections. One Health 2023, 16, 100518. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serovar | No. of Isolates | No. of Outbreaks | cgMLST Allele Range | 7-Gene MLST | Vehicle Category (Vehicle) |
|---|---|---|---|---|---|
| Adelaide | 2 | 1 | 3 | 440 | Meat (pork) |
| Agbeni | 24 | 1 | 0–21 | 2606, ND | Zoonotic (turtles) |
| Altona | 4 | 1 | 1–4 | 1549 | Zoonotic (live poultry) |
| Bareilly | 4 | 1 | 0–2 | 909 | Fish (raw tuna scrape) |
| Berta | 3 | 1 | 5–7 | 435 | Poultry (ground turkey) |
| Bovismorbificans | 2 | 1 | 2 | 377 | Seeds (hummus) |
| Braenderup | 16 | 3 | 1–2 | 22, ND | Zoonotic (live poultry) |
| 0–2 | 22 | Zoonotic (live poultry) | |||
| 0–6 | 22 | Zoonotic (live poultry) | |||
| Bredeney | 4 | 1 | 1–4 | 505 | Nuts (peanut butter) |
| Carrau | 4 | 1 | 0–2 | 226 | Fruit (melon) |
| Chester | 5 | 1 | 0–2 | 2063 | Multiple (frozen dinners) |
| Cubana | 4 | 1 | 2–6 | 286 | Vegetable (sprouts) |
| Enteritidis | 28 | 7 | 0 | 11 | Zoonotic (reptile/live rodent) |
| 2–3 | 11 | Vegetable (sprouts) | |||
| 0–2 | 11 | Nuts (Turkish pine nuts) | |||
| 1–6 | 11 | Vegetable (lettuce) | |||
| 3–6 | 11 | Fish (tuna sushi) | |||
| 0 | 11 | Poultry (chicken) | |||
| 0 | 11 | Poultry (breaded stuffed chicken) | |||
| Hadar | 23 | 4 | 1–2 | 33 | Poultry (turkey) |
| 0 | 33 | Poultry (ground turkey) | |||
| 0–5 | 33 | Zoonotic (live poultry) | |||
| 0–4 | 33 | Zoonotic (live poultry) | |||
| Hartford | 2 | 1 | 2 | 405 | Vegetable (salsa) |
| Heidelberg | 18 | 5 | 0 | 15 | Seeds (hummus) |
| 0–2 | 15 | Poultry (ground turkey) | |||
| 0 | 15 | Eggs (custard-filled pastry) | |||
| 1–5 | 15 | Poultry (ground turkey) | |||
| 0 | 15 | Poultry (chicken liver) | |||
| I 4,[5],12:i:- | 29 | 5 | 0–4 | 19 | Multiple (pot pies) |
| 10–22 | 19 | Poultry (breaded stuffed chicken) | |||
| 0–10 | 19 | Pet food (frozen rodents) | |||
| 4–14 | 19 | Zoonotic (rodents) | |||
| 2–4 | 34 | Vegetable (lettuce) | |||
| Infantis | 10 | 3 | 3–5 | 32 | Pet food (dog food) |
| 0 | 32 | Zoonotic (live poultry) | |||
| 0 | 32 | Meat (pork) | |||
| Javiana | 58 | 5 | 0–1 | 24 | Vegetable (tomatoes) |
| 0 | 24 | Vegetable (tomatoes) | |||
| 0–1 | 24 | Vegetable (cucumbers) | |||
| 0–3 | 24 | Fish (tilapia) | |||
| 0–4 | 24 | Multiple (potato salad) | |||
| Johannesburg | 7 | 2 | 0–1 | 471 | Zoonotic (live poultry) |
| 0–5 | 471 | Zoonotic (live poultry) | |||
| Kiambu | 3 | 1 | 0–2 | 309 | Zoonotic (live poultry) |
| Litchfield | 5 | 1 | 0–1 | 214 | Fruit (cantaloupe melon) |
| Montevideo | 15 | 6 | 7 | 138, ND | Dairy (shredded cheese) |
| 1 | 316 | Poultry (unspecified) | |||
| 0 | 81 | Meat (pork) | |||
| 4 | 4 | Zoonotic (live poultry) | |||
| 2 | 4 | Zoonotic (live poultry) | |||
| 0–5 | 316 | Zoonotic (live poultry) | |||
| Muenchen | 7 | 2 | 0–2 | 112 | Fruit (blueberries) |
| 3–5 | 83 | Zoonotic (live poultry) | |||
| Newport | 66 | 17 | 20 | 118 | Vegetable (tomatoes) |
| 15 | 45 | Dairy (queso fresco-type cheeses) | |||
| 5–13 | 118 | Vegetable (tomatoes) | |||
| 0 | 118 | Meat (beef) | |||
| 0–2 | 2362, ND | Vegetable (green chilies) | |||
| 0 | 45 | Vegetable (lettuce) | |||
| 1–2 | 45 | Meat (ground beef) | |||
| 0–1 | 45 | Vegetable (tomatoes) | |||
| 0–1 | 118 | Vegetable (tomatoes) | |||
| 0–1 | 118 | Fruit (cantaloupe) | |||
| 1–3 | 45 | Vegetable (tomatoes) | |||
| 0–7 | 118 | Fruit (cantaloupe melon) | |||
| 0 | 5 | Vegetable (romaine lettuce) | |||
| 0–2 | 45 | Meat (beef) | |||
| 0–2 | 118 | Dairy (queso fresco-type cheeses) | |||
| 0 | 45 | Dairy (queso fresco-type cheeses) | |||
| 0 | 45 | Meat (pork) | |||
| Paratyphi B var. L(+) tartrate+ | 8 | 3 | 0–1 | 43 | Fish (ahi) |
| 0 | 43 | Multiple (raw tuna) | |||
| 0 | 307 | Multiple (raw fish) | |||
| Pomona | 3 | 1 | 2 | 451 | Zoonotic (turtles) |
| Poona | 2 | 1 | 2 | 3095 | Zoonotic (turtles) |
| Rissen | 4 | 1 | 0–5 | 469 | Vegetable (white/black pepper) |
| Saintpaul | 7 | 2 | 1–2 | 50 | Multiple (steak, spice rub) |
| 2–5 | 50 | Vegetable (cucumbers) | |||
| Sandiego | 5 | 2 | 6–13 | 20 | Zoonotic (turtles) |
| 4 | 126 | Zoonotic (turtles) | |||
| Schwarzengrund | 7 | 2 | 2–11 | 96 | Zoonotic (live poultry market) |
| 2–8 | 96 | Zoonotic (live poultry market) | |||
| Senftenberg | 3 | 1 | 0–1 | 185 | Nuts (pistachios) |
| Tennessee | 5 | 1 | 0–18 | 319 | Nuts (peanut butter) |
| Thompson | 3 | 1 | 1–2 | 26 | Zoonotic (live poultry) |
| Typhimurium | 48 | 13 | 1–3 | 19 | Vegetable (tomatoes) |
| 1–2 | 19 | Water (municipal water) | |||
| 1–3 | 19 | Vegetable (sprouts) | |||
| 2 | 19, ND | Meat (ground beef) | |||
| 4 | 19 | Zoonotic (live poultry) | |||
| 0–1 | 19 | Fruit (cantaloupe melon) | |||
| 1–3 | 19 | Fruit (cantaloupe melon) | |||
| 0–5 | 19 | Zoonotic (hedgehogs) | |||
| 0–2 | 19 | Zoonotic (turtles) | |||
| 0 | 19 | Fruit (unpasteurized apple cider) | |||
| 1–2 | 19 | Zoonotic (live poultry) | |||
| 0 | 19 | Meat (ground beef) | |||
| 0–5 | 19 | Zoonotic (live poultry) | |||
| Total (35) | 438 | 100 |
| PulseNet Cluster Code * | Serovar | Vehicle | No. of Isolates | cgMLST Allele Range | hqSNP Range |
|---|---|---|---|---|---|
| 0608PAJJP-1 ** | Newport | Tomatoes | 2 | 20 | 47 |
| 0611ILJJP-1 | Newport | Queso fresco-type cheeses | 2 | 15 | 24 |
| 0708NYJJP-1 ** | Newport | Tomatoes | 3 | 5–13 | 11–38 |
| 0802COJIX-1 | Montevideo | Shredded cheese | 2 | 7 | 10 |
| 0809MLJPX-1 ** | I 4,[5],12:i:- | Breaded stuffed chicken | 3 | 10–22 | 24–46 |
| 0907MLJM6-1 | Schwarzengrund | Live poultry contact | 3 | 2–11 | 7–16 |
| 1508MLJLX-1 | Sandiego | Live turtle contact | 3 | 6–13 | 7–18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trees, E.; Carleton, H.A.; Folster, J.P.; Gieraltowski, L.; Hise, K.; Leeper, M.; Nguyen, T.-A.; Poates, A.; Sabol, A.; Tagg, K.A.; et al. Genetic Diversity in Salmonella enterica in Outbreaks of Foodborne and Zoonotic Origin in the USA in 2006–2017. Microorganisms 2024, 12, 1563. https://doi.org/10.3390/microorganisms12081563
Trees E, Carleton HA, Folster JP, Gieraltowski L, Hise K, Leeper M, Nguyen T-A, Poates A, Sabol A, Tagg KA, et al. Genetic Diversity in Salmonella enterica in Outbreaks of Foodborne and Zoonotic Origin in the USA in 2006–2017. Microorganisms. 2024; 12(8):1563. https://doi.org/10.3390/microorganisms12081563
Chicago/Turabian StyleTrees, Eija, Heather A. Carleton, Jason P. Folster, Laura Gieraltowski, Kelley Hise, Molly Leeper, Thai-An Nguyen, Angela Poates, Ashley Sabol, Kaitlin A. Tagg, and et al. 2024. "Genetic Diversity in Salmonella enterica in Outbreaks of Foodborne and Zoonotic Origin in the USA in 2006–2017" Microorganisms 12, no. 8: 1563. https://doi.org/10.3390/microorganisms12081563
APA StyleTrees, E., Carleton, H. A., Folster, J. P., Gieraltowski, L., Hise, K., Leeper, M., Nguyen, T.-A., Poates, A., Sabol, A., Tagg, K. A., Tolar, B., Vasser, M., Webb, H. E., Wise, M., & Lindsey, R. L. (2024). Genetic Diversity in Salmonella enterica in Outbreaks of Foodborne and Zoonotic Origin in the USA in 2006–2017. Microorganisms, 12(8), 1563. https://doi.org/10.3390/microorganisms12081563