
Microbial Biogeography along the Gastrointestinal Tract of a Wild Chinese Muntjac (Muntiacus reevesi)

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. DNA Extraction, PCR Amplification and Illumina MiSeq Sequencing
2.4. Data Analysis
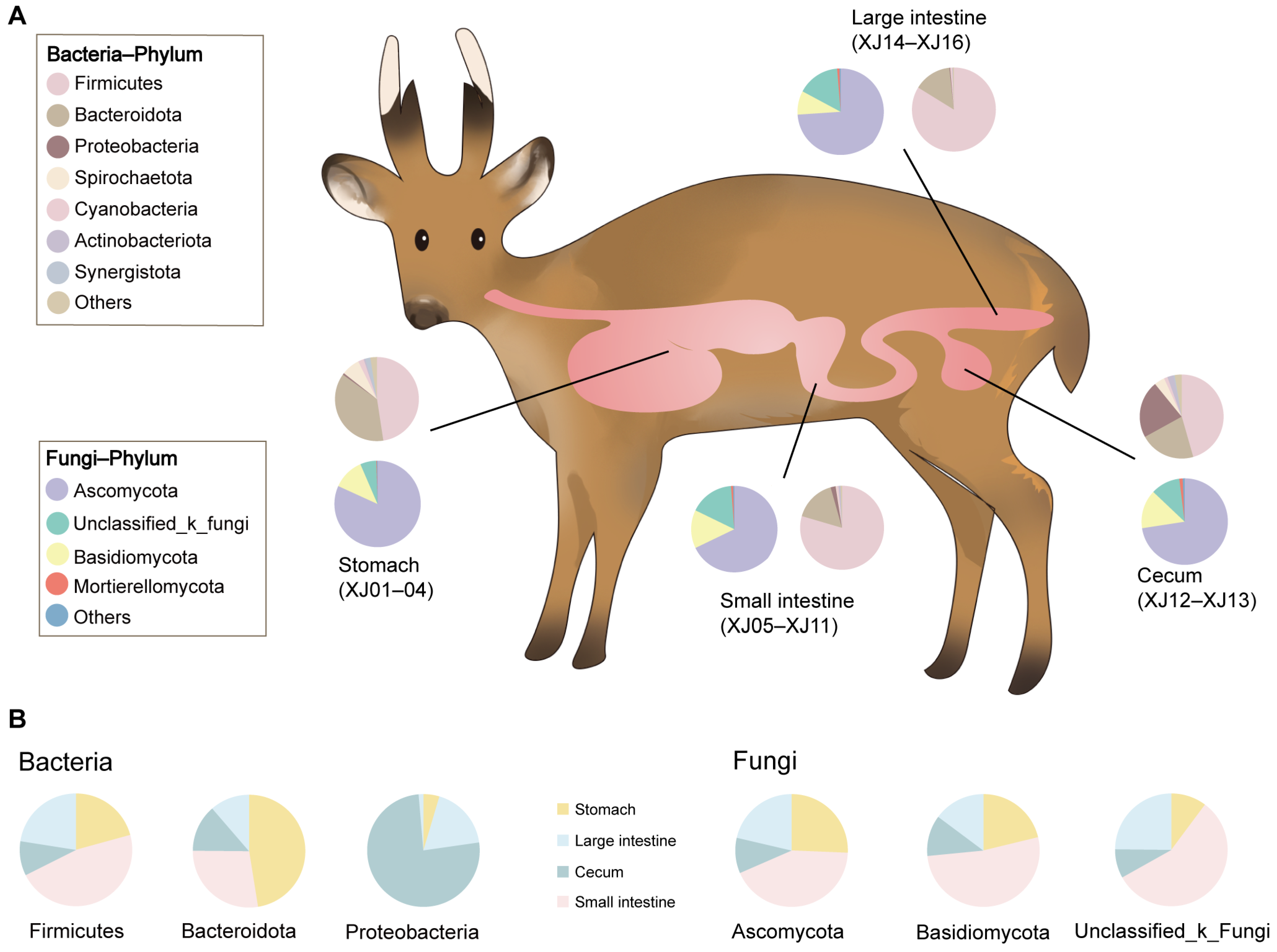
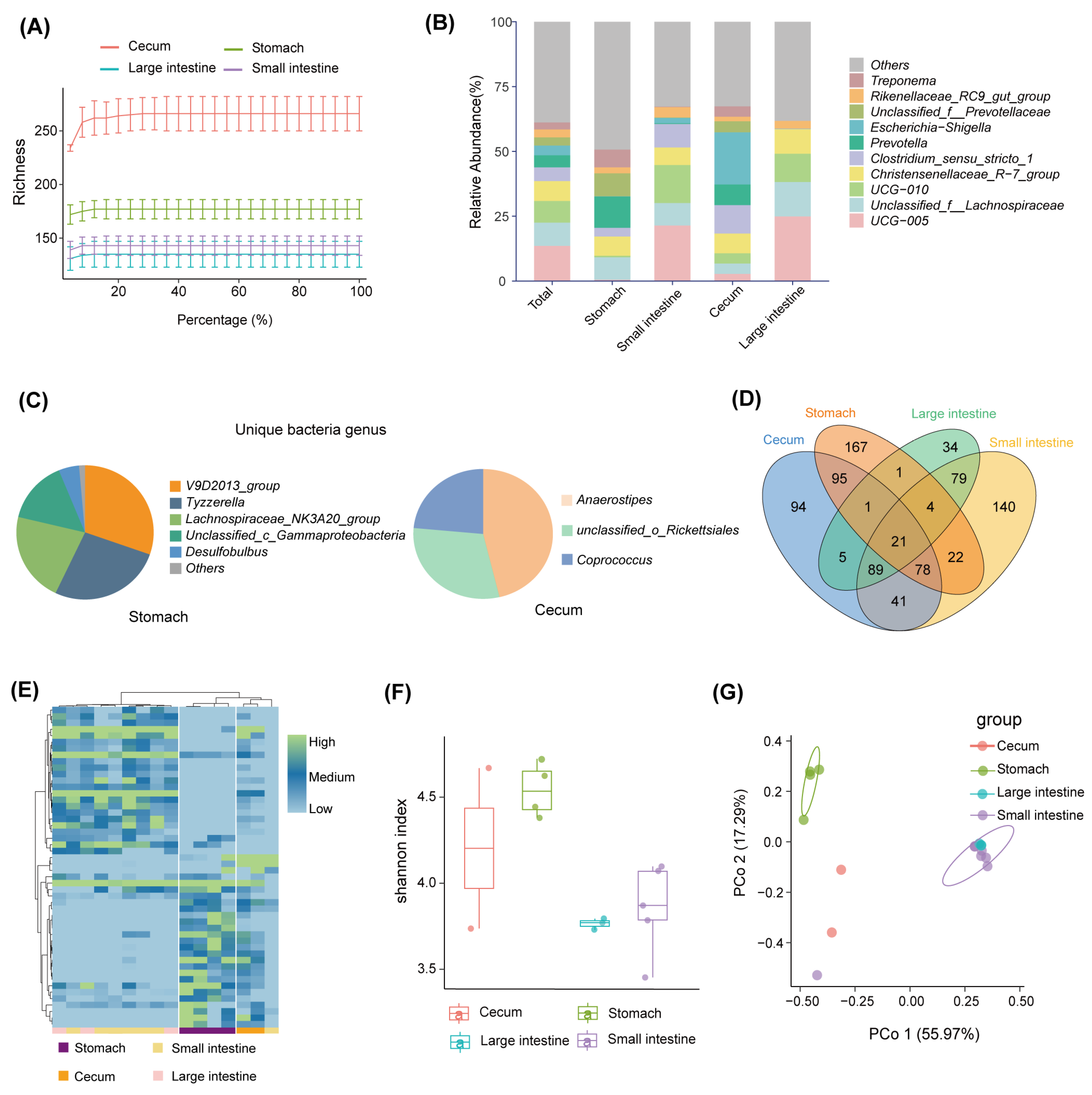
3. Results
3.1. Bacterial Community Diversity
3.2. Fungal Community Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Escalas, A.; Hale, L.; Voordeckers, J.W.; Yang, Y.; Firestone, M.K.; Alvarez-Cohen, L.; Zhou, J. Microbial functional diversity: From concepts to applications. Ecol. Evol. 2019, 9, 12000–12016. [Google Scholar] [CrossRef]
- An, C.; Okamoto, Y.; Xu, S.; Eo, K.Y.; Kimura, J.; Yamamoto, N. Comparison of fecal microbiota of three captive carnivore species inhabiting Korea. J. Vet. Med. Sci. 2017, 79, 542–546. [Google Scholar] [CrossRef]
- Huang, G.; Wang, X.; Hu, Y.; Wu, Q.; Nie, Y.; Dong, J.; Ding, Y.; Yan, L.; Wei, F. Diet drives convergent evolution of gut microbiomes in bamboo-eating species. Sci. China Life Sci. 2021, 64, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Fruciano, C.; Hildebrand, F.; Al Toufalilia, H.; Balfour, N.J.; Bork, P.; Engel, P.; Ratnieks, F.L.; Hughes, W.O. Gut microbiota composition is associated with environmental landscape in honey bees. Ecol. Evol. 2018, 8, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Romanenko, M.; Piven, L.; Moseiko, V.; Lushchak, O.; Kryzhanovska, N.; Guryanov, V.; Koliada, A. Differences in the gut firmicutes to bacteroidetes ratio across age groups in healthy Ukrainian population. BMC Microbiol. 2020, 20, 221. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Handl, S.; Dowd, S.E.; Garcia-Mazcorro, J.F.; Steiner, J.M.; Suchodolski, J.S. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol. Ecol. 2011, 76, 301–310. [Google Scholar] [CrossRef]
- Kohl, K.D.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.C.; Caviedes-Vidal, E.; Bordenstein, S.R. Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 2017, 26, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial Community Mapping of the Mouse Gastrointestinal Tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.J.; Gillett, A.; Polkinghorne, A.; Timms, P. Investigation of the koala (Phascolarctos cinereus) hindgut microbiome via 16S pyrosequencing. Vet. Microbiol. 2013, 167, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wei, Y.; Zhang, T.; Wang, X.; Xu, Y.; Zhang, W.; Zheng, Y. Gastrointestinal Biogeography of Luminal Microbiota and Short-Chain Fatty Acids in Sika Deer (Cervus nippon). Appl. Environ. Microbiol. 2022, 88, e00499-22. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, Q.; Yu, J.; Wang, K.; Lin, Z.S.; Li, Z.P.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef] [PubMed]
- Sbardellati, D.L.; Fischer, A.; Cox, M.S.; Li, W.; Kalscheur, K.F.; Suen, G. The bovine epimural microbiota displays compositional and structural heterogeneity across different ruminal locations. J. Dairy Sci. 2020, 103, 3636–3647. [Google Scholar] [CrossRef]
- O’ Donnell, M.M.; Harris, H.M.B.; Ross, R.P.; O’Toole, P.W. Core fecal microbiota of domesticated herbivorous ruminant, hindgut fermenters, and monogastric animals. Microbiol. Open 2017, 6, e00509. [Google Scholar] [CrossRef]
- Hu, X.; Liu, G.; Li, Y.; Wei, Y.; Lin, S.; Liu, S.; Zheng, Y.; Hu, D. High-Throughput Analysis Reveals Seasonal Variation of the Gut Microbiota Composition Within Forest Musk Deer (Moschus berezovskii). Front. Microbiol. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Li, J.; Wang, C.; Tang, Z.; Guo, Y.; Zheng, T.; Li, Y.; You, Z. The Gut Bacterial Community Composition of Wild Cervus albirostris (White-Lipped Deer) Detected by the 16S Ribosomal RNA Gene Sequencing. Curr. Microbiol. 2017, 74, 1100–1107. [Google Scholar] [CrossRef]
- Yan, W.; Sun, C.; Zheng, J.; Wen, C.; Ji, C.; Zhang, D.; Chen, Y.; Hou, Z.; Yang, N. Efficacy of Fecal Sampling as a Gut Proxy in the Study of Chicken Gut Microbiota. Front. Microbiol. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Sun, Z.; Orozco-terWengel, P.; Chen, G.; Sun, R.; Sun, L.; Wang, H.; Shi, W.; Zhang, B. Spatial dynamics of Chinese Muntjac related to past and future climate fluctuations. Curr. Zool. 2021, 67, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic. Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, I.; Wallace, R.J.; Moraïs, S. The rumen microbiome: Balancing food security and environmental impacts. Nat. Rev. Microbiol. 2021, 19, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hong, S.W.; Park, B.Y.; Yoo, J.G.; Oh, M.H. Characterization of the bacterial community in the gastrointestinal tracts of elk (Cervus canadensis). Antonie Van Leeuwenhoek 2019, 112, 225–235. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Xu, C.; Zhao, J.; Liu, H.; Fan, Z.; Yang, F.; Wright, A.-D.G.; Li, G. Bacteria and methanogens differ along the gastrointestinal tract of Chinese roe deer (Capreolus pygargus). PLoS ONE 2014, 9, e114513. [Google Scholar] [CrossRef]
- Li, Z.; Wright, A.-D.G.; Liu, H.; Bao, K.; Zhang, T.; Wang, K.; Cui, X.; Yang, F.; Zhang, Z.; Li, G. Bacterial community composition and fermentation patterns in the rumen of sika deer (Cervus nippon) fed three different diets. Microb. Ecol 2015, 69, 307–318. [Google Scholar] [CrossRef]
- Waite, D.W.; Taylor, M.W. Characterizing the avian gut microbiota: Membership, driving influences, and potential function. Front. Microbiol. 2014, 5, 223. [Google Scholar] [CrossRef]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, L.; Xue, B.; Wang, Z.; Peng, Q. Characterizing the bacterial community across the gastrointestinal tract of goats: Composition and potential function. Microbiol. Open 2019, 8, e00820. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.K.; Do, T.H.; Le, N.G.; Nguyen, H.D.; Nguyen, T.Q.; Le, T.T.H.; Truong, N.H. Understanding the role of prevotella genus in the digestion of lignocellulose and other substrates in vietnamese native goats’ rumen by metagenomic deep sequencing. Animals 2021, 11, 3257. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xu, L.; Wang, Y.; Mao, S. Metagenomic sequencing reveals that high-grain feeding alters the composition and metabolism of cecal microbiota and induces cecal mucosal injury in sheep. Msystems 2021, 6, e0091521. [Google Scholar] [CrossRef] [PubMed]
- Guerra, V.; Tiago, I.; Aires, A.; Coelho, C.; Nunes, J.; Martins, L.O.; Veríssimo, A. The gastrointestinal microbiome of browsing goats (Capra hircus). PLoS ONE 2022, 17, e0276262. [Google Scholar] [CrossRef]
- Yang, C.; Tsedan, G.; Liu, Y.; Hou, F. Shrub coverage alters the rumen bacterial community of yaks (Bos grunniens) grazing in alpine meadows. J. Anim. Sci. Technol. 2020, 62, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ. 2017, 32, 300–313. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian Australas. J. Anim. Sci 2017, 30, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Ji, Z.; Shen, Z.; Xue, Y.; Zhang, B.; Yu, D.; Liu, T.; Luo, D.; Xing, G.; Tang, J.; et al. Increase Dietary Fiber Intake Ameliorates Cecal Morphology and Drives Cecal Species-Specific of Short-Chain Fatty Acids in White Pekin Ducks. Front. Microbiol. 2022, 13, 853797. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.H. Butyrate producers, “The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2022, 13, 1103836. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.L.; McLaughlin, R.W.; Zheng, J.S.; Hao, Y.J.; Fan, F.; Tian, R.M.; Wang, D. Microbial communities in different regions of the gastrointestinal tract in East Asian finless porpoises (Neophocaena asiaeorientalis sunameri). Sci. Rep. 2018, 8, 14142. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.B.; Meng, J.X.; Ma, H.; Liu, R.; Qin, Y.; Qin, Y.F.; Geng, H.L.; Ni, H.B.; Zhang, X.X. Description of Gut Mycobiota Composition and Diversity of Caprinae Animals. Microbiol. Spectr. 2023, 11, e0242422. [Google Scholar] [CrossRef] [PubMed]
- Aira, M.; Pérez-Losada, M.; Crandall, K.A.; Domínguez, J. Host taxonomy determines the composition, structure, and diversity of the earthworm cast microbiome under homogenous feeding conditions. FEMS Microbiol. Ecol. 2022, 98, 1–10. [Google Scholar] [CrossRef]
- Cholewińska, P.; Górniak, W.; Wojnarowski, K. Impact of selected environmental factors on microbiome of the digestive tract of ruminants. BMC Vet. Res. 2021, 17, 25. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Salonen, A.; Vos, W.M. Impact of Diet on Human Intestinal Microbiota and Health. Annu. Rev. Food Sci. Technol. 2014, 5, 239–262. [Google Scholar] [CrossRef]
- Brown, K.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Removal of the cecum affects intestinal fermentation, enteric bacterial community structure, and acute colitis in mice. Gut Microbes 2018, 9, 218–235. [Google Scholar] [CrossRef]
- Krehbiel, C.R. Applied nutrition of ruminants: Fermentation and digestive physiology. Prof. Anim. Sci 2014, 30, 129–139. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Shu, Y.; Huang, Y.; Tan, J.; Wang, F.; Tang, L.; Fang, T.; Yuan, S.; Wang, L. Microbial Biogeography along the Gastrointestinal Tract of a Wild Chinese Muntjac (Muntiacus reevesi). Microorganisms 2024, 12, 1587. https://doi.org/10.3390/microorganisms12081587
Liu Y, Shu Y, Huang Y, Tan J, Wang F, Tang L, Fang T, Yuan S, Wang L. Microbial Biogeography along the Gastrointestinal Tract of a Wild Chinese Muntjac (Muntiacus reevesi). Microorganisms. 2024; 12(8):1587. https://doi.org/10.3390/microorganisms12081587
Chicago/Turabian StyleLiu, Yuan, Yan Shu, Yuling Huang, Jinchao Tan, Fengmei Wang, Lin Tang, Tingting Fang, Shibin Yuan, and Le Wang. 2024. "Microbial Biogeography along the Gastrointestinal Tract of a Wild Chinese Muntjac (Muntiacus reevesi)" Microorganisms 12, no. 8: 1587. https://doi.org/10.3390/microorganisms12081587
APA StyleLiu, Y., Shu, Y., Huang, Y., Tan, J., Wang, F., Tang, L., Fang, T., Yuan, S., & Wang, L. (2024). Microbial Biogeography along the Gastrointestinal Tract of a Wild Chinese Muntjac (Muntiacus reevesi). Microorganisms, 12(8), 1587. https://doi.org/10.3390/microorganisms12081587