
Bacterial Communities Associated with the Leaves and the Roots of Salt Marsh Plants of Bayfront Beach, Mobile, Alabama, USA

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sample Collection and Preservation
2.2. DNA Sample Preparation
2.3. Library Construction and High-Throughput DNA Sequencing of 16S rDNA
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
2.6. Microbial Network Construction and Analysis
3. Results
3.1. Microbial Community Composition and Diversity
3.2. Shared and Unique ASVs and Keystone Taxa
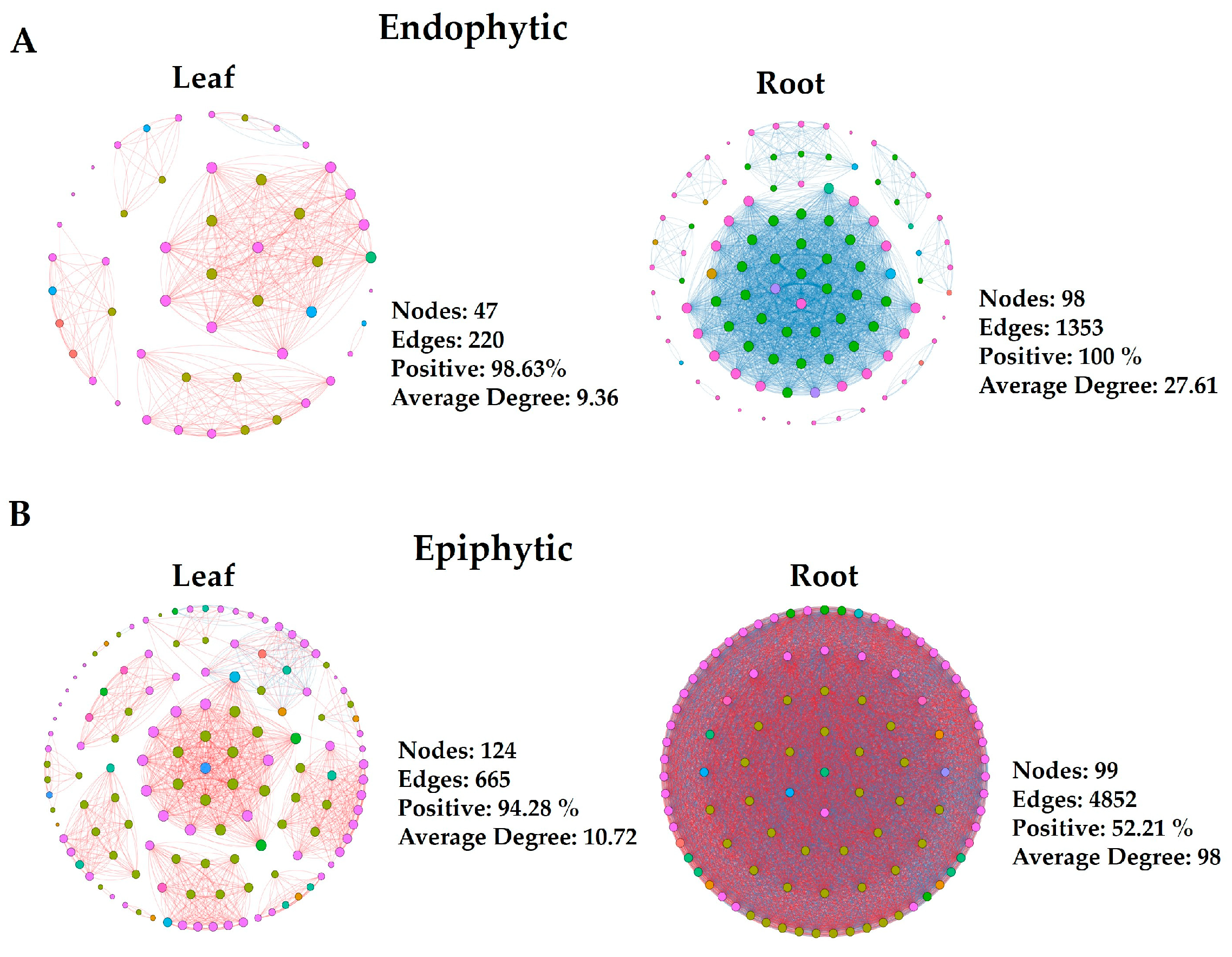
3.3. Microbial Co-Occurrence Network
3.4. Bacterial Diversity Analyses Differentiate Communities among Habitats
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mcowen, C.J.; Weatherdon, L.V.; Van Bochove, J.-W.; Sullivan, E.; Blyth, S.; Zockler, C.; Stanwell-Smith, D.; Kingston, N.; Martin, C.S.; Spalding, M. A global map of saltmarshes. Biodivers. Data J. 2017, 5, e11764. [Google Scholar] [CrossRef] [PubMed]
- Rolando, J.L.; Kolton, M.; Song, T.; Kostka, J.E. The core root microbiome of Spartina alterniflora is predominated by sulfur-oxidizing and sulfate-reducing bacteria in Georgia salt marshes, USA. Microbiome 2022, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Silliman, B.R.; Cui, B. Incorporating thresholds into understanding salinity tolerance: A study using salt-tolerant plants in salt marshes. Ecol. Evol. 2017, 7, 6326–6333. [Google Scholar] [CrossRef] [PubMed]
- Soldan, R.; Mapelli, F.; Crotti, E.; Schnell, S.; Daffonchio, D.; Marasco, R.; Fusi, M.; Borin, S.; Cardinale, M. Bacterial endophytes of mangrove propagules elicit early establishment of the natural host and promote growth of cereal crops under salt stress. Microbiol. Res. 2019, 223, 33–43. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Grube, M.; Köberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2016, 67, 995–1002. [Google Scholar] [CrossRef]
- Zheng, Y.; Xu, Z.; Liu, H.; Liu, Y.; Zhou, Y.; Meng, C.; Ma, S.; Xie, Z.; Li, Y.; Zhang, C.-S. Patterns in the microbial community of salt-tolerant plants and the functional genes associated with salt stress alleviation. Microbiol. Spectr. 2021, 9, e0076721. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef]
- Yang, X.; Dai, Z.; Yuan, R.; Guo, Z.; Xi, H.; He, Z.; Wei, M. Effects of salinity on assembly characteristics and function of microbial communities in the phyllosphere and rhizosphere of salt-tolerant Avicennia marina mangrove species. Microbiol. Spectr. 2023, 11, e03000–e03022. [Google Scholar] [CrossRef]
- Guo, J.; Chen, Y.; Lu, P.; Liu, M.; Sun, P.; Zhang, Z. Roles of endophytic bacteria in Suaeda salsa grown in coastal wetlands: Plant growth characteristics and salt tolerance mechanisms. Environ. Pollut. 2021, 287, 117641. [Google Scholar] [CrossRef]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Qu, X.; Pan, Y.; Wang, P.; Ran, L.; Qin, G.; Li, Q.; Kang, P. Response of Phyllosphere and Rhizosphere Microbial Communities to Salt Stress of Tamarix chinensis. Plants 2024, 13, 1091. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jang, Y.-J.; Lee, S.-M.; Oh, B.-T.; Chae, J.-C.; Lee, K.-J. Alleviation of salt stress by Enterobacter sp. EJ01 in tomato and Arabidopsis is accompanied by up-regulation of conserved salinity responsive factors in plants. Mol. Cells 2014, 37, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Paul, S.C.; Parveen, S.; Alam, S.; Rahman, N.; Jannat, B.; Hoque, S.; Rahman, M.T.; Karim, M.M. Isolation and identification of salt-tolerant plant-growth-promoting rhizobacteria and their application for rice cultivation under salt stress. Can. J. Microbiol. 2020, 66, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Bano, A. Isolation of plant-growth-promoting rhizobacteria from rhizospheric soil of halophytes and their impact on maize (Zea mays L.) under induced soil salinity. Can. J. Microbiol. 2015, 61, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, H.; Naeem, S.; Bakhtawar, M.; Jabeen, Z.; Nosheen, A.; Naz, R.; Keyani, R.; Mumtaz, S.; Hassan, M.N. Halotolerant rhizobacteria Pseudomonas pseudoalcaligenes and Bacillus subtilis mediate systemic tolerance in hydroponically grown soybean (Glycine max L.) against salinity stress. PLoS ONE 2020, 15, e0231348. [Google Scholar] [CrossRef] [PubMed]
- Travis, S.E.; Simon, M.R.; Zogg, G.P. Environmental Sensitivity of Soil Microbial Communities is Altered in Association with Plant Roots in Saltmarsh Ecosystems. Northeast. Nat. 2024, 31, 146–162. [Google Scholar] [CrossRef]
- Jiang, H.; Okoye, C.O.; Chen, X.; Zhang, F.; Jiang, J. High-throughput 16S rRNA gene-based amplicon sequencing reveals the functional divergence of halophilic bacterial communities in the Suaeda salsa root compartments on the Eastern Coast of China. Sci. Total Environ. 2024, 942, 173775. [Google Scholar] [CrossRef] [PubMed]
- Rush, G.I.; Clark, B.; Lumibao, C.Y. Biotic versus abiotic factors shaping culturable root endosymbionts of the saltmarsh halophyte, Batis maritima and implications for plant stress tolerance. Wetl. Ecol. Manag. 2024, 32, 453–461. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2018, 2, 1–295. [Google Scholar]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Xie, P.; Yang, S.; Niu, G.; Liu, X.; Ding, Z.; Xue, C.; Liu, Y.X.; Shen, Q.; Yuan, J. ggClusterNet: An R package for microbiome network analysis and modularity-based multiple network layouts. Imeta 2022, 1, e32. [Google Scholar] [CrossRef]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef]
- Mateus, M.; Almeida, D.; Simonson, W.; Felgueiras, M.; Banza, P.; Batty, L. Conflictive uses of coastal areas: A case study in a southern European coastal lagoon (Ria de Alvor, Portugal). Ocean Coast. Manag. 2016, 132, 90–100. [Google Scholar] [CrossRef]
- Zheng, W.; Xue, D.; Li, X.; Deng, Y.; Rui, J.; Feng, K.; Wang, Z.-L. The responses and adaptations of microbial communities to salinity in farmland soils: A molecular ecological network analysis. Appl. Soil Ecol. 2017, 120, 239–246. [Google Scholar] [CrossRef]
- Szymańska, S.; Borruso, L.; Brusetti, L.; Hulisz, P.; Furtado, B.; Hrynkiewicz, K. Bacterial microbiome of root-associated endophytes of Salicornia europaea in correspondence to different levels of salinity. Environ. Sci. Pollut. Res. 2018, 25, 25420–25431. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, M.; Tan, J.; Zhang, C.; Liu, Y.; Huang, J.; Tan, Y.; Xiao, L.; Xu, Z. Salt-tolerant plant moderates the effect of salinity on soil organic carbon mineralization in a subtropical tidal wetland. Sci. Total Environ. 2022, 837, 155855. [Google Scholar] [CrossRef]
- Lenk, S.; Arnds, J.; Zerjatke, K.; Musat, N.; Amann, R.; Mußmann, M. Novel groups of Gammaproteobacteria catalyse sulfur oxidation and carbon fixation in a coastal, intertidal sediment. Environ. Microbiol. 2011, 13, 758–774. [Google Scholar] [CrossRef]
- Wang, K.; Wang, S.; Zhang, X.; Wang, W.; Li, F.; Dong, L.; Kong, F.; Xi, M. Potential ecological impacts of physical control on Spartina alterniflora in coastal wetland: Migration and transformation of nutrients and the response of bacterial community structure. J. Clean. Prod. 2023, 398, 136556. [Google Scholar] [CrossRef]
- Kim, C.; Staver, L.W.; Chen, X.; Bulseco, A.; Cornwell, J.C.; Malkin, S.Y. Microbial community succession along a chronosequence in constructed salt marsh soils. Microb. Ecol. 2023, 85, 931–950. [Google Scholar] [CrossRef]
- An, X.; Wang, Z.; Teng, X.; Zhou, R.; Wang, X.; Xu, M.; Lian, B. Rhizosphere bacterial diversity and environmental function prediction of wild salt-tolerant plants in coastal silt soil. Ecol. Indic. 2022, 134, 108503. [Google Scholar] [CrossRef]
- Petrosyan, K.; Thijs, S.; Piwowarczyk, R.; Ruraż, K.; Kaca, W.; Vangronsveld, J. Diversity and potential plant growth promoting capacity of seed endophytic bacteria of the holoparasite Cistanche phelypaea (Orobanchaceae). Sci. Rep. 2023, 13, 11835. [Google Scholar] [CrossRef] [PubMed]
- Doni, F.; Suhaimi, N.S.M.; Irawan, B.; Mohamed, Z.; Mispan, M.S. Associations of Pantoea with rice plants: As friends or foes? Agriculture 2021, 11, 1278. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, L.; Sun, H.; Gao, M.; Yu, N.; Zhang, Q.; Mou, A.; Liu, Y. Microbial co-occurrence network topological properties link with reactor parameters and reveal importance of low-abundance genera. NPJ Biofilms Microbiomes 2022, 8, 3. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majeed, A.; Liu, J.; Knight, A.J.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. Bacterial Communities Associated with the Leaves and the Roots of Salt Marsh Plants of Bayfront Beach, Mobile, Alabama, USA. Microorganisms 2024, 12, 1595. https://doi.org/10.3390/microorganisms12081595
Majeed A, Liu J, Knight AJ, Pajerowska-Mukhtar KM, Mukhtar MS. Bacterial Communities Associated with the Leaves and the Roots of Salt Marsh Plants of Bayfront Beach, Mobile, Alabama, USA. Microorganisms. 2024; 12(8):1595. https://doi.org/10.3390/microorganisms12081595
Chicago/Turabian StyleMajeed, Aqsa, Jinbao Liu, Adelle J. Knight, Karolina M. Pajerowska-Mukhtar, and M. Shahid Mukhtar. 2024. "Bacterial Communities Associated with the Leaves and the Roots of Salt Marsh Plants of Bayfront Beach, Mobile, Alabama, USA" Microorganisms 12, no. 8: 1595. https://doi.org/10.3390/microorganisms12081595