
Salmonirosea aquatica gen. nov., sp. nov., a Novel Genus within the Family Spirosomaceae, Was Isolated from Brackish Water in the Republic of Korea


Abstract
:1. Introduction
2. Material and Methods
2.1. Isolation and Ecology
2.2. 16S rRNA Gene Phylogeny
2.3. Genome Sequencing, Assembly and Annotation
2.4. Morphological and Phenotypic Characterization
2.5. Chemotaxonomic Characterization
3. Results and Discussion
3.1. Isolation of Strain
3.2. 16S rRNA Gene Phylogeny
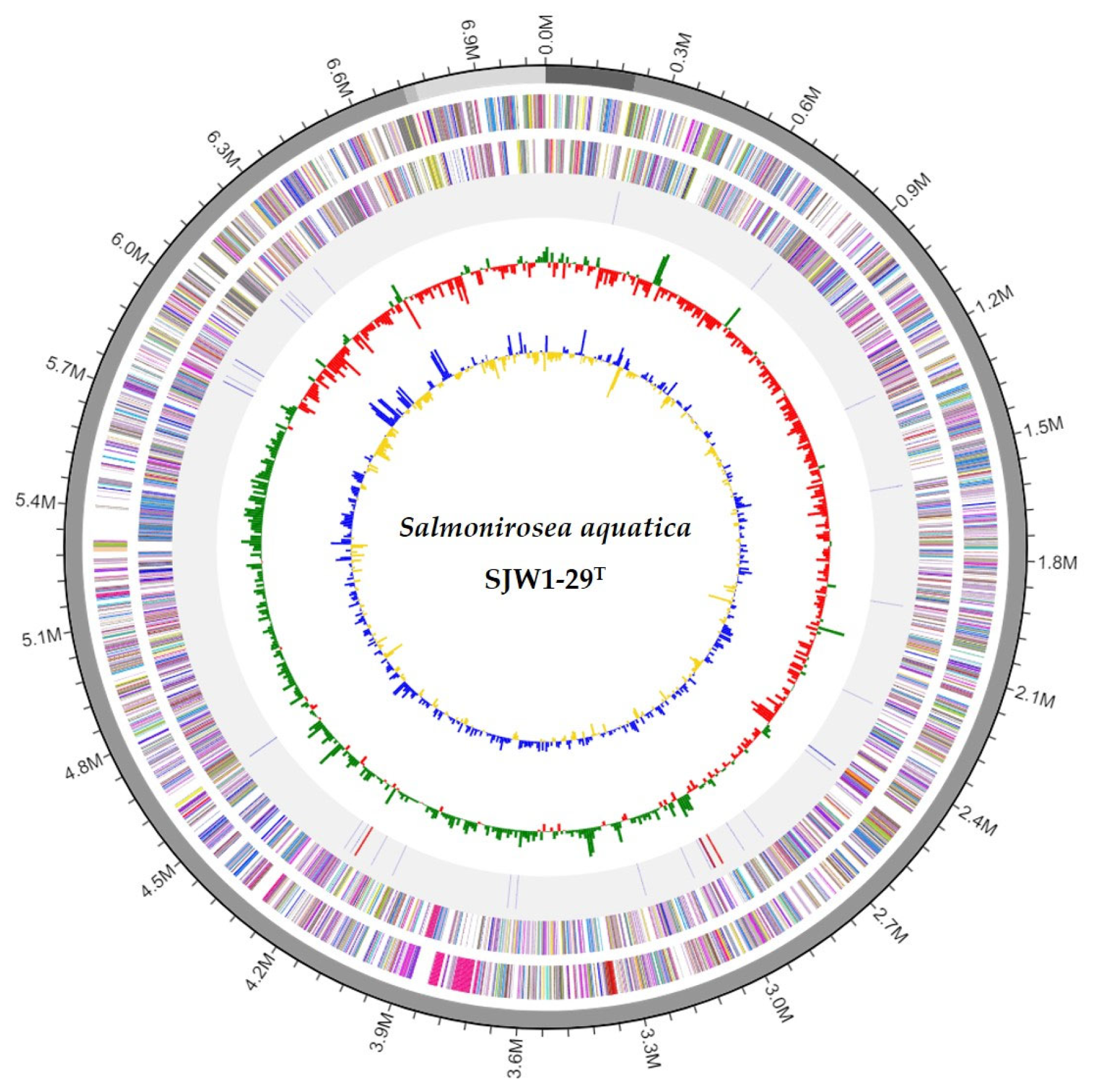
3.3. Genome Features
3.4. Morphological and Phenotypic Characteristics

{kind=link}
{kind=link}
{kind=link}
| Characteristic | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Isolation source | Brackish water | Sea water | Soil | Freshwater | Soil | Plant | Soil |
| 16S rRNA gene similarity (%) | ref. | 91.3 | 90.6 | 90.0 | 89.1 | 88.4 | 84.8 |
| Genome size (bp) | 7,065,248 | 6,413,589 | 6,917,964 | 5,828,278 | 6,558,731 | 6,967,790 | 5,775,379 |
| ANI value | ref. | 75.0 | 71.9 | 71.8 | 71.1 | 69.2 | 67.3 |
| AAI value | ref. | 78.4 | 70.8 | 74.6 | 70.6 | 67.0 | 63.4 |
| dDDH value | ref. | 21.0 | 18.5 | 17.5 | 18.2 | 18.8 | 19.9 |
| POCP value | ref. | 62.6 | 63.9 | 66.0 | 63.0 | 52.6 | 46.0 |
| Colony color | Salmon pink | Pale pink | Light pink | Light pink | Pink | Yellow | Pale yellow |
| Cell morphology | Rods | Straight rod to curved rods | Rods | Rods | Rods | Rods | Rods |
| Flexirubin reaction | − | − | ND | ND | ND | + | ND |
| Temperature for growth (°C) | 15–30 | 25–30 | 10–37 | 5–37 | 10–42 | 15–37 | 15–40 |
| Highest NaCl tolerated (%, w/v) | <5.0 | <7.0 | <5.0 | <4.0 | <4.0 | <1.5 | <1.0 |
| Genome size (Mb) | 7.07 | ND | ND | 5.83 | ND | 6.97 | ND |
| Assimilation of (API 20NE): | |||||||
| Glucose fermentation | − | + | − | W | + | + | − |
| Urease | − | + | − | + | ND | ND | + |
| Enzyme activity (API ZYM) | |||||||
| Alkaline phosphatase | + | − | − | + | + | ND | + |
| Esterase (C4), cystine arylamidase, trypsin, β-galactosidase | − | + | + | + | + | ND | + |
| Lipase (C14) | − | − | − | − | − | ND | + |
| α-chymotrypsin | + | + | + | − | + | ND | − |
| Acid phosphatase | + | + | + | + | − | ND | + |
| β-glucosidase | + | + | + | − | + | ND | + |
| α-mannosidase | + | − | + | + | + | ND | + |
| Major quinone † | MK-7 | MK-7 | MK-7 | MK-7 | MK-7 | MK-7 | MK-7 |
| Major polar lipids ‡ | PE, APL, AL, L | PE, AL, L | PE, PL, AL, L | PE, AL, GL, L | AL, L | PE, PL, AL, L | PE, APL, AL |
| DNA G+C content (mol%) § | 50.7 | 50.8 | 54.0 | 45.7 | 48.9 | 51.5 | 50.6 |
3.5. Chemotaxonomic Characterization
4. Conclusions
4.1. Description of Salmonirosea gen. nov.
4.2. Description of Salmonirosea aquatica sp. nov.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Larkin, J.M.; Borrall, R. Spirosomaceae, a new family to contain the genera Spirosoma Migula 1894, Flectobacillus Larkin et al. 1977, and Runella Larkin and Williams 1978. Int. J. Syst. Evol. Microbiol. 1978, 28, 595–596. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G. Notification of changes in taxonomic opinion previously published outside the IJSEM. Int. J. Syst. Evol. Microbiol. 2020, 70, 4061–4090. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.L.; Ruberto, L.; Calabro, A.; Balbo, A.L.; Del Panno, M.T.; Mac Cormack, W.P. Hydrocarbon removal and bacterial community structure in on-site biostimulated biopile systems designed for bioremediation of diesel-contaminated Antarctic soil. Polar Biol. 2015, 38, 677–687. [Google Scholar] [CrossRef]
- El Fantroussi, S.; Agathos, S.N. Is bioaugmentation a feasible strategy for pollutant removal and site remediation? Curr. Opin. Microbiol. 2005, 8, 268–275. [Google Scholar] [CrossRef]
- Dixit, R.; Wasiullah; Malaviya, D.; Pandiyan, K.; Singh, U.B.; Sahu, A.; Shukla, R.; Singh, B.P.; Rai, J.P.; Sharma, P.K.; et al. Bioremediation of heavy metals from soil and aquatic environment: An overview of principles and criteria of fundamental processes. Sustainability 2015, 7, 2189–2212. [Google Scholar] [CrossRef]
- Donati, E.R.; Sani, R.K.; Goh, K.M.; Chan, K.-G. Editorial: Recent advances in bioremediation/biodegradation by extreme microorganisms. Front. Microbiol. 2019, 10, 1851. [Google Scholar] [CrossRef]
- Khan, F.I.; Husain, T.; Hejazi, R. An overview and analysis of site remediation technologies. J. Environ. Manag. 2004, 71, 95–122. [Google Scholar] [CrossRef]
- Hatti-Kaul, R.; Törnvall, U.; Gustafsson, L.; Börjesson, P. Industrial biotechnology for the production of bio-based chemicals—A cradle-to-grave perspective. Trends Biotechnol. 2007, 25, 119–124. [Google Scholar] [CrossRef]
- Duarte, M.; Nielsen, A.; Camarinha-Silva, A.; Vilchez-Vargas, R.; Bruls, T.; Wos-Oxley, M.L.; Jauregui, R.; Pieper, D.H. Functional soil metagenomics: Elucidation of polycyclic aromatic hydrocarbon degradation potential following 12 years of in situ bioremediation. Environ. Microbiol. 2017, 19, 2992–3011. [Google Scholar] [CrossRef] [PubMed]
- Olaniran, A.O.; Pillay, D.; Pillay, B. Biostimulation and bioaugmentation enhances aerobic biodegradation of dichloroethenes. Chemosphere 2006, 63, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Halim, M.; Conte, P.; Piccolo, A. Potential availability of heavy metals to phytoextraction from contaminated soils induced by exogenous humic substances. Chemosphere 2003, 52, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Kumar, V.; Pal, A.K. Microbial degradation of high molecular weight polycyclic aromatic hydrocarbons with emphasis on pyrene. Polycycl. Aromat. Compd. 2019, 39, 124–138. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, M.J.; LEE, G.G. National level assessment of Biodiversity importance—Focusing on South Korea. KSCE J. Civ. Eng. 2014, 19, 46–62. [Google Scholar] [CrossRef]
- Lee, M.; Par, B.S.; Baek, S.H. Tidal Influences on Biotic and Abiotic Factors in the Seomjin River Estuary and Gwangyang Bay, Korea. Estuaries Coast 2018, 41, 1977–1993. [Google Scholar] [CrossRef]
- Hwang, J.H.; Jang, D.; Kim, Y.H. Stratification and salt-wedge in the Seomjin river estuary under the idealized tidal influence. Ocean Sci. J. 2017, 52, 469–487. [Google Scholar] [CrossRef]
- Prieto-Barajas, C.M.; Valencia-Cantero, E.; Santoyo, G. Microbial mat ecosystems: Structure types, functional diversity, and biotechnological application. Electron. J. Biotechnol. 2018, 31, 48–56. [Google Scholar] [CrossRef]
- Lee, D.I.; Park, C.K.; Cho, H.S. Ecological modeling for water quality management of Kwangyang Bay, Korea. J. Environ. Manag. 2005, 74, 327–337. [Google Scholar] [CrossRef]
- Findlay, S.J.; Taylor, M.P. Why rehabilitate urban river systems? Area 2006, 38, 312–325. [Google Scholar] [CrossRef]
- Gurnell, A.; Lee, M.; Souch, C. Urban rivers: Hydrology, geomorphology, ecology and opportunities for change. Geogr. Compass 2007, 1, 1118–1137. [Google Scholar] [CrossRef]
- Paul, M.J.; Meyer, J.L. Streams in the urban landscape. Annu. Rev. Ecol. Syst. 2001, 32, 333–365. [Google Scholar] [CrossRef]
- Bunn, S.E.; Arthington, A.H. Basic principles and ecological consequences of altered flow regimes for aquatic biodiversity. Environ. Manag. 2002, 30, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.S.; Lee, K.; Park, S.C.; Kim, B.S.; Cho, Y.J.; Ha, S.M.; Chun, J. EzEditor: A versatile sequence alignment editor for both rRNA- and protein-coding genes. Int. J. Syst. Evol. Microbiol. 2014, 64, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Chalita, M.; Ha, S.M.; Na, S.I.; Yoon, S.H.; Chun, J. ContEst16S: An algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2017, 67, 2053–2057. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A pipeline for high throughput calculations of prokaryotic average amino acid identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef]
- Choo, Y.-J.; Lee, K.; Song, J.; Cho, J.-C. Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales ord. nov., Opitutaceae fam. nov., Opitutales ord. nov. and Opitutae classis nov. in the phylum ‘Verrucomicrobia’. Int. J. Syst. Evol. Microbiol. 2007, 57, 532–537. [Google Scholar] [CrossRef]
- Smibert, R.M.; Krieg, N.R. Phenotypic characterization. In Methods for General and Molecular Bacteriology; Gerhardt, P., Murray, R.G.E., Wood, W.A., Krieg, N.R., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 607–654. [Google Scholar]
- Tindall, B.J.; Sikorski, J.; Smibert, R.A.; Krieg, N.R. Phenotypic Characterization and the Principles of Comparative Systematics. In Methods for General and Molecular Microbiology; Reddy, C.A., Ed.; ASM Press: Washington, DC, USA, 2007; pp. 330–393. [Google Scholar]
- Sasser, M. Identification of Bacteria by Gas Chromatography of Cellular Fatty Acids; Technical Note 101; MIDI: Newark, NJ, USA, 2001. [Google Scholar]
- Minnikin, D.E.; O’Donnell, A.G.; Goodfellow, M.; Alderson, G.; Athalye, M.; Schaal, A.; Parlett, J.H. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 1984, 2, 233–241. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Baek, K.; Choi, A. Draft genome sequence of the polysaccharide degrading bacterium Cytophagaceae strain SJW1-29. Korean J. Microbiol. 2020, 56, 422–425. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Rosselló-Móra, R.; Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017, 11, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B.M. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Hölzer, M. POCP-nf: An automatic Nextflow pipeline for calculating the percentage of conserved proteins in bacterial taxonomy. Bioinformatics 2024, 40, btae175. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, H.; Lebre, P.; Blom, J.; Cowan, D.; De Maayer, P. Phylogenomic re-assessment of the thermophilic genus Geobacillus. Syst. Appl. Microbiol. 2016, 39, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Ishikawa, S.; Kasai, H.; Yokota, A. Persicitalea jodogahamensis gen. nov., sp. nov., a marine bacterium of the family ‘Flexibacteraceae’, isolated from seawater in Japan. Int. J. Syst. Evol. Microbiol. 2007, 57, 1014–1017. [Google Scholar] [CrossRef]
- Dahal, R.H.; Kim, J. Rhabdobacter roseus gen. nov., sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 2016, 66, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Szuróczki, S.; Khayer, B.; Spröer, C.; Toumi, M.; Szabó, A.; Felföldi, T.; Schumann, P.; Tóth, E. Arundinibacter roseus gen. nov., sp. nov., a new member of the family Cytophagaceae. Int. J. Syst. Evol. Microbiol. 2019, 69, 2076–2081. [Google Scholar] [CrossRef]
- Lee, D.; Jang, J.H.; Cha, S.; Seo, T. Telluribacter humicola gen. nov., sp. nov., a new member of the family Cytophagaceae isolated from soil in South Korea. Antonie Van Leeuwenhoek 2016, 109, 1525–1533. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. Dyadobacter fermentans gen. nov., sp. nov., a novel gram-negative bacterium isolated from surface-sterilized Zea mays stems. Int. J. Syst. Evol. Microbiol. 2000, 50, 751–758. [Google Scholar] [CrossRef]
- Chaudhary, D.K.; Dahal, R.H.; Altankhuu, K.; Kim, J. Ravibacter arvi gen. nov., sp. nov., isolated from farmland soil during development of new culture techniques. Int. J. Syst. Evol. Microbiol. 2017, 67, 5252–5260. [Google Scholar] [CrossRef] [PubMed]


| Fatty Acid | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Saturated | |||||||
| C14:0 | 1.9 | - | 1.0 | - | tr | - | tr |
| C16:0 | 8.0 | 12.0 | 6.6 | 7.4 | 7.7 | 5.6 | 10.1 |
| Unsaturated | |||||||
| C13:1 | - | 2.0 | 1.6 | - | - | 1.3 | - |
| C14:1 ω5c | - | - | - | - | 3.7 | - | |
| C16:1 ω5c | 1.8 | 6.6 | 17.5 | 14.2 | 13.5 | 15.9 | 6.9 |
| Hydroxy | |||||||
| C14:0 2-OH | 1.1 | - | - | - | - | - | tr |
| C16:0 3-OH | 1.2 | 2.2 | 1.3 | 2.3 | - | 2.3 | 4.6 |
| C16:0 N alcohol | - | - | - | - | 1.1 | - | - |
| Iso-C15:0 3-OH | 2.6 | 2.1 | 1.0 | 1.5 | 3.2 | 1.7 | 2.5 |
| Iso-C16:0 2-OH | - | - | - | - | - | - | 1.1 |
| Iso-C16:0 3-OH | 2.6 | 1.5 | 1.2 | - | - | - | tr |
| Iso-C17:0 3-OH | 8.2 | 6.2 | 3.7 | 6.5 | - | 4.4 | 8.5 |
| Iso-C17:1 ω9c | - | - | - | 1.3 | 3.9 | - | - |
| Branched chain | |||||||
| Iso-C10:0 | - | - | - | - | 4.3 | - | - |
| Iso-C14:0 | 1.1 | - | - | - | - | - | tr |
| Iso-C15:0 | 11.9 | 31.5 | 21.4 | 16.6 | 13.8 | 19.4 | 9.9 |
| Iso-C17:0 | - | - | - | - | 1.8 | - | tr |
| Iso-C15:0 G | - | - | - | 1.4 | - | - | |
| Anteiso-C15:0 | 1.5 | 1.7 | - | 1.7 | 3.5 | - | tr |
| Summed features 3 * | 55.3 | 29.9 | 40.7 | 41.8 | 39.9 | 44.4 | 52.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, K.; Jang, S.; Goh, J.; Choi, A. Salmonirosea aquatica gen. nov., sp. nov., a Novel Genus within the Family Spirosomaceae, Was Isolated from Brackish Water in the Republic of Korea. Microorganisms 2024, 12, 1671. https://doi.org/10.3390/microorganisms12081671
Baek K, Jang S, Goh J, Choi A. Salmonirosea aquatica gen. nov., sp. nov., a Novel Genus within the Family Spirosomaceae, Was Isolated from Brackish Water in the Republic of Korea. Microorganisms. 2024; 12(8):1671. https://doi.org/10.3390/microorganisms12081671
Chicago/Turabian StyleBaek, Kiwoon, Sumin Jang, Jaeduk Goh, and Ahyoung Choi. 2024. "Salmonirosea aquatica gen. nov., sp. nov., a Novel Genus within the Family Spirosomaceae, Was Isolated from Brackish Water in the Republic of Korea" Microorganisms 12, no. 8: 1671. https://doi.org/10.3390/microorganisms12081671