

Adaptive Evolution Signatures in Prochlorococcus: Open Reading Frame (ORF)eome Resources and Insights from Comparative Genomics

 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Protein Family Domain Prediction
2.2. Hierarchical Bi-Clustering
2.3. Response Screening
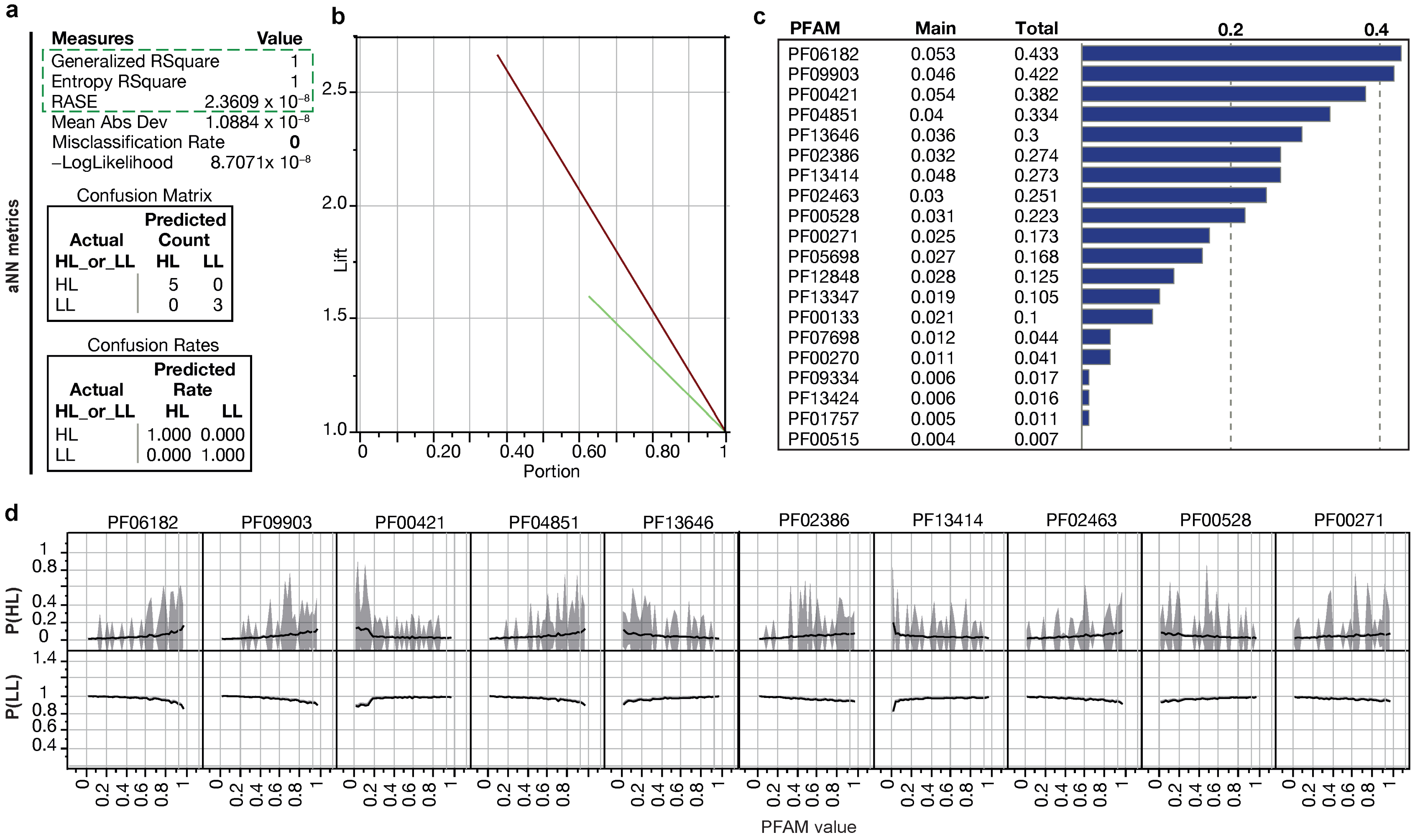
2.4. Deep Learning ANN Analysis
2.5. VFam Analysis
2.6. Metabolic Pathway Analysis
2.7. Functional Annotation Analysis Using Blast2GO
2.8. ORFeome Synthesis and Cloning
3. Results
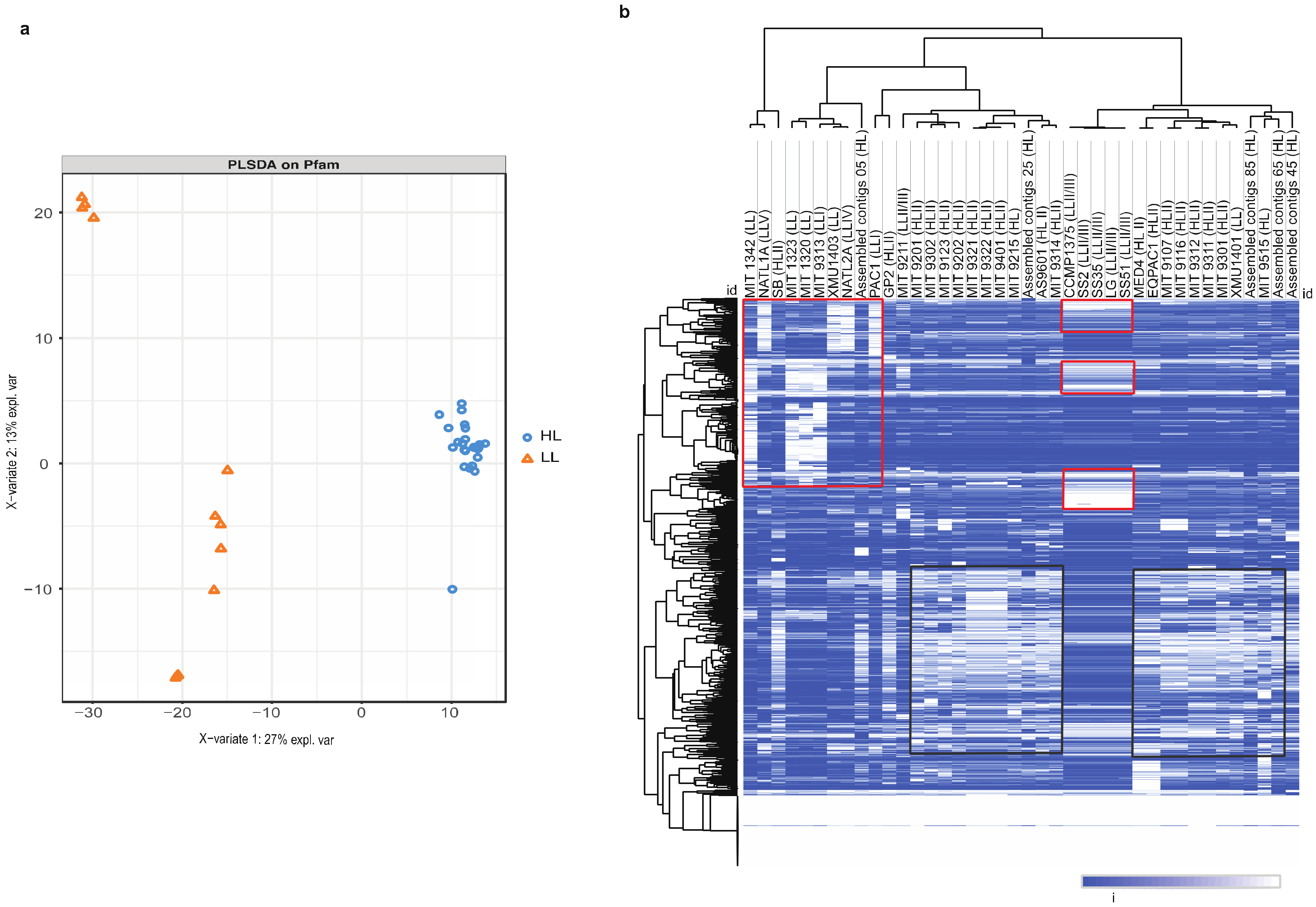
3.1. Decoding High-Light and Low-Light Associated Gene Sets from P. marinus Genomes
3.2. Identification of Minimal Pfam Sets Distinguishing HL and LL Strains
3.3. Variable, Depth-Dependent Accumulation of Endogenous Viral Elements
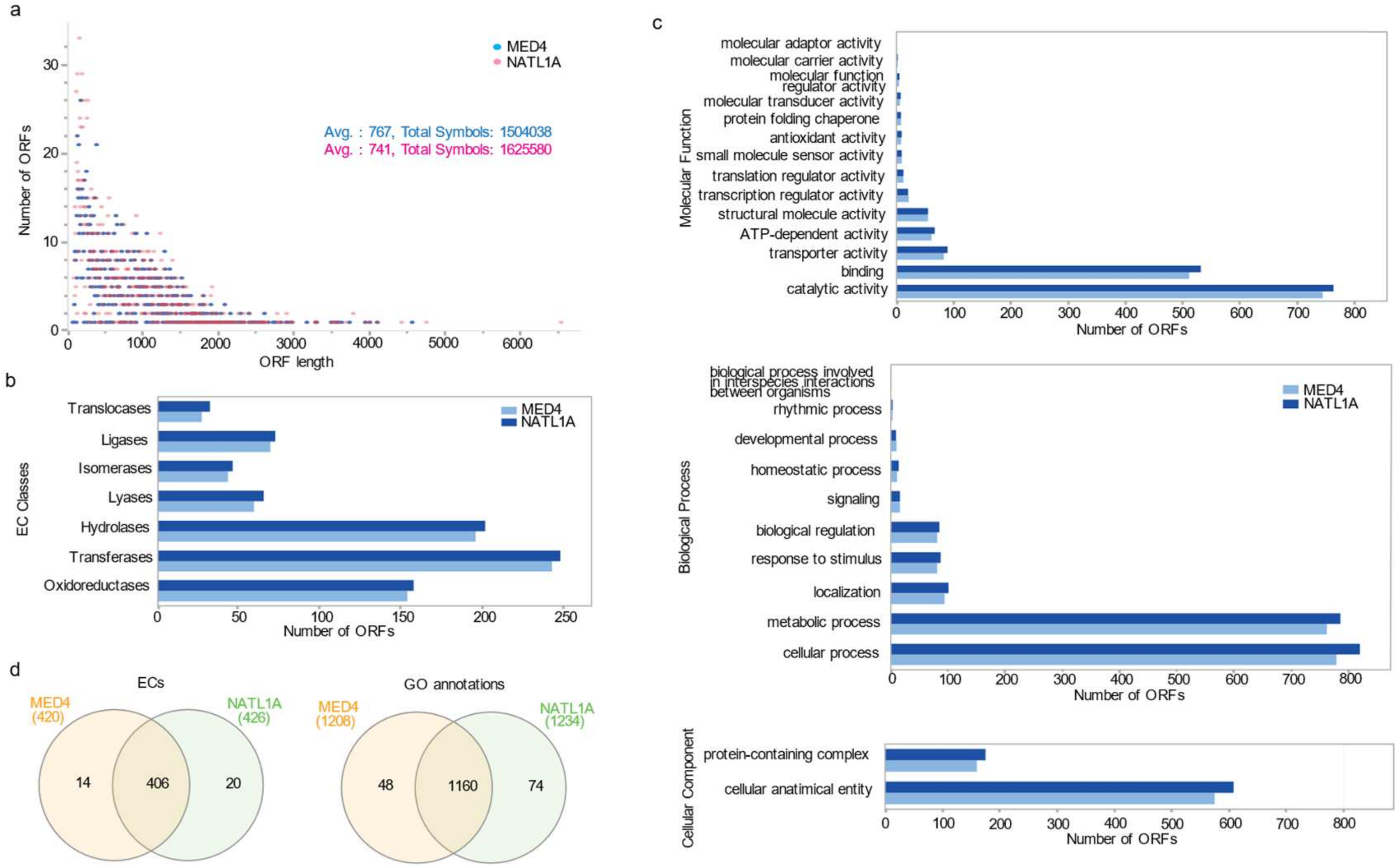
3.4. The Prochlorococcus Strains MED4 and NATL1A as HL and LL Representatives
3.5. ORFeome Resource Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Percival, S.L.; Williams, D.W. Microbiology of Waterborne Diseases, 2nd ed.; Percival, S.L., Yates, M.V., Williams, D.W., Chalmers, R.M., Gray, N.F., Eds.; Academic Press: London, UK, 2014; pp. 79–88. [Google Scholar]
- Demoulin, C.F.; Lara, Y.J.; Cornet, L.; François, C.; Baurain, D.; Wilmotte, A.; Javaux, E.J. Cyanobacteria evolution: Insight from the fossil record. Free Radic. Biol. Med. 2019, 140, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Ruffing, A.M. Engineered cyanobacteria: Teaching an old bug new tricks. Bioeng. Bugs 2011, 2, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Goericke, R.; Welschmeyer, N.A. The marine prochlorophyte Prochlorococcus contributes significantly to phytoplankton biomass and primary production in the Sargasso Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 1993, 40, 2283–2294. [Google Scholar] [CrossRef]
- Scanlan, D.J.; Ostrowski, M.; Mazard, S.; Dufresne, A.; Garczarek, L.; Hess, W.R.; Post, A.F.; Hagemann, M.; Paulsen, I.; Partensky, F. Ecological genomics of marine picocyanobacteria. Microbiol. Mol. Biol. Rev. 2009, 73, 249–299. [Google Scholar] [CrossRef] [PubMed]
- Partensky, F.; Hess, W.R.; Vaulot, D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 1999, 63, 106. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Rocap, G.; Chisholm, S.W. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature 1998, 393, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Huang, Y.; Stepanauskas, R.; Tang, J. Excess of non-conservative amino acid changes in marine bacterioplankton lineages with reduced genomes. Nat. Microbiol. 2017, 2, 17091. [Google Scholar] [CrossRef] [PubMed]
- Kettler, G.C.; Martiny, A.C.; Huang, K.; Zucker, J.; Coleman, M.L.; Rodrigue, S.; Chen, F.; Lapidus, A.; Ferriera, S.; Johnson, J.; et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 2007, 3, e231. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A.; Salanoubat, M.; Partensky, F.; Artiguenave, F.; Axmann, I.M.; Barbe, V.; Duprat, S.; Galperin, M.Y.; Koonin, E.V.; Le Gall, F.; et al. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc. Natl. Acad. Sci. USA 2003, 100, 10020–10025. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Goericke, R.; Chisholm, S.W. Comparative physiology of Synechococcus and Prochlorococcus: Influence of light and temperature on growth, pigments, fluorescence and absorptive properties. Mar. Ecol. Prog. Ser. 1995, 116, 259–275. [Google Scholar] [CrossRef]
- Rocap, G.; Larimer, F.W.; Lamerdin, J.; Malfatti, S.; Chain, P.; Ahlgren, N.A.; Arellano, A.; Coleman, M.; Hauser, L.; Hess, W.R.; et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 2003, 424, 1042–1047. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Rual, J.-F.; Hirozane-Kishikawa, T.; Hao, T.; Bertin, N.; Li, S.; Dricot, A.; Li, N.; Rosenberg, J.; Lamesch, P.; Vidalain, P.-O.; et al. Human ORFeome Version 1.1: A Platform for Reverse Proteomics. Genome Res. 2004, 14, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Darzi, Y.; Letunic, I.; Bork, P.; Yamada, T. iPath3.0: Interactive pathways explorer v3. Nucleic Acids Res. 2018, 46, W510–W513. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4, 17. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Teng, W.-K.; Zhao, L.; Hu, C.-X.; Zhou, Y.-K.; Han, B.-P.; Song, L.-R.; Shu, W.-S. Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J. 2021, 15, 211–227. [Google Scholar] [CrossRef]
- Yang, F.; Miao, Y.; Liu, Y.; Botella, J.R.; Li, W.; Li, K.; Song, C.-P. Function of Protein Kinases in Leaf Senescence of Plants. Front. Plant Sci. 2022, 13, 864215. [Google Scholar] [CrossRef] [PubMed]
- Andrés, G.; María, F.F.; María-Teresa, B.; María-Luisa, P.; Emma, S. Cyanobacteria; Archana, T., Ed.; IntechOpen: Rijeka, Croatia, 2018; Chapter 6. [Google Scholar]
- Jia, A.; Zheng, Y.; Chen, H.; Wang, Q. Regulation and Functional Complexity of the Chlorophyll-Binding Protein IsiA. Front. Microbiol. 2021, 12, 774107. [Google Scholar] [CrossRef] [PubMed]
- Kroh, G.E.; Pilon, M. Regulation of iron homeostasis and use in chloroplasts. Int. J. Mol. Sci. 2020, 21, 3395. [Google Scholar] [CrossRef]
- Christensen, Q.H.; Cronan, J.E. Lipoic acid synthesis: A new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry 2010, 49, 10024–10036. [Google Scholar] [CrossRef]
- Cronan, J.E. Assembly of Lipoic Acid on Its Cognate Enzymes: An Extraordinary and Essential Biosynthetic Pathway. Microbiol. Mol. Biol. Rev. 2016, 80, 429–450. [Google Scholar] [CrossRef]
- Bristow, L.A.; Mohr, W.; Ahmerkamp, S.; Kuypers, M.M.M. Nutrients that limit growth in the ocean. Curr. Biol. 2017, 27, R474–R478. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Ruiz, M.; Zhang, C.-C. Oxidative stress in cyanobacteria. FEMS Microbiol. Rev. 2009, 33, 258–278. [Google Scholar] [CrossRef]
- Santamaría-Gómez, J.; Ochoa de Alda, J.A.G.; Olmedo-Verd, E.; Bru-Martínez, R.; Luque, I. Sub-Cellular Localization and Complex Formation by Aminoacyl-tRNA Synthetases in Cyanobacteria: Evidence for Interaction of Membrane-Anchored ValRS with ATP Synthase. Front. Microbiol. 2016, 7, 857. [Google Scholar] [CrossRef]
- Luque, I.; Riera-Alberola, M.L.; Andújar, A.; Ochoa de Alda, J.A.G. Intraphylum Diversity and Complex Evolution of Cyanobacterial Aminoacyl-tRNA Synthetases. Mol. Biol. Evol. 2008, 25, 2369–2389. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Baumgartner, D.; Hagemann, M.; Muro-Pastor, A.M.; Maaß, S.; Becher, D.; Hess, W.R. AtpΘ is an inhibitor of F0F1 ATP synthase to arrest ATP hydrolysis during low-energy conditions in cyanobacteria. Curr. Biol. 2022, 32, 136–148.e135. [Google Scholar] [CrossRef]
- Cassier-Chauvat, C.; Veaudor, T.; Chauvat, F. Comparative Genomics of DNA Recombination and Repair in Cyanobacteria: Biotechnological Implications. Front. Microbiol. 2016, 7, 1809. [Google Scholar] [CrossRef] [PubMed]
- Kolowrat, C.; Partensky, F.; Mella-Flores, D.; Le Corguillé, G.; Boutte, C.; Blot, N.; Ratin, M.; Ferréol, M.; Lecomte, X.; Gourvil, P.; et al. Ultraviolet stress delays chromosome replication in light/dark synchronized cells of the marine cyanobacterium Prochlorococcus marinus PCC9511. BMC Microbiol. 2010, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Osburne, M.S.; Holmbeck, B.M.; Frias-Lopez, J.; Steen, R.; Huang, K.; Kelly, L.; Coe, A.; Waraska, K.; Gagne, A.; Chisholm, S.W. UV hyper-resistance in Prochlorococcus MED4 results from a single base pair deletion just upstream of an operon encoding nudix hydrolase and photolyase. Environ. Microbiol. 2010, 12, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Cassier-Chauvat, C.; Chauvat, F. Responses to oxidative and heavy metal stresses in cyanobacteria: Recent advances. Int. J. Mol. Sci. 2014, 16, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Berube, P.M.; Rasmussen, A.; Braakman, R.; Stepanauskas, R.; Chisholm, S.W. Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. eLife 2019, 8, e41043. [Google Scholar] [CrossRef]
- Varkey, D.; Mazard, S.; Ostrowski, M.; Tetu, S.G.; Haynes, P.; Paulsen, I.T. Effects of low temperature on tropical and temperate isolates of marine Synechococcus. ISME J. 2016, 10, 1252–1263. [Google Scholar] [CrossRef]
- Knoll, A.; Puchta, H. The role of DNA helicases and their interaction partners in genome stability and meiotic recombination in plants. J. Exp. Bot. 2010, 62, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, M.; Karow, A.R.; Klostermeier, D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol. Chem. 2009, 390, 1237–1250. [Google Scholar] [CrossRef]
- Muzzopappa, F.; Wilson, A.; Yogarajah, V.; Cot, S.; Perreau, F.; Montigny, C.; De Carbon, C.B.; Kirilovsky, D. Paralogs of the C-Terminal Domain of the Cyanobacterial Orange Carotenoid Protein Are Carotenoid Donors to Helical Carotenoid Proteins. Plant Physiol. 2017, 175, 1283–1303. [Google Scholar] [CrossRef] [PubMed]
- Cerveny, L.; Straskova, A.; Dankova, V.; Hartlova, A.; Ceckova, M.; Staud, F.; Stulik, J. Tetratricopeptide repeat motifs in the world of bacterial pathogens: Role in virulence mechanisms. Infect. Immun. 2013, 81, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Grove, T.Z.; Cortajarena, A.L.; Regan, L. Ligand binding by repeat proteins: Natural and designed. Curr. Opin. Struct. Biol. 2008, 18, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Rast, A.; Rengstl, B.; Heinz, S.; Klingl, A.; Nickelsen, J. The Role of Slr0151, a Tetratricopeptide Repeat Protein from Synechocystis sp. PCC 6803, during Photosystem II Assembly and Repair. Front. Plant Sci. 2016, 7, 605. [Google Scholar] [CrossRef] [PubMed]
- Biller, S.J.; Coe, A.; Chisholm, S.W. Torn apart and reunited: Impact of a heterotroph on the transcriptome of Prochlorococcus. ISME J. 2016, 10, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Xu, X.; Hu, Z. A TPR-family membrane protein gene is required for light-activated heterotrophic growth of the cyanobacterium Synechocystis sp. PCC 6803. FEMS Microbiol. Lett. 2003, 219, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Nishio, K.; Nakai, M. Identification of a novel prokaryotic HEAT-repeats-containing protein which interacts with a cyanobacterial IscA homolog. FEBS Lett. 2002, 519, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Mella-Flores, D.; Six, C.; Ratin, M.; Partensky, F.; Boutte, C.; Le Corguillé, G.; Marie, D.; Blot, N.; Gourvil, P.; Kolowrat, C.; et al. Prochlorococcus and Synechococcus have Evolved Different Adaptive Mechanisms to Cope with Light and UV Stress. Front. Microbiol. 2012, 3, 285. [Google Scholar] [CrossRef] [PubMed]
- Ofaim, S.; Sulheim, S.; Almaas, E.; Sher, D.; Segrè, D. Dynamic Allocation of Carbon Storage and Nutrient-Dependent Exudation in a Revised Genome-Scale Model of Prochlorococcus. Front. Genet. 2021, 12, 586293. [Google Scholar] [CrossRef] [PubMed]
- Bibby, T.S.; Mary, I.; Nield, J.; Partensky, F.; Barber, J. Low-light-adapted Prochlorococcus species possess specific antennae for each photosystem. Nature 2003, 424, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Gisriel, C.J.; Shen, G.; Flesher, D.A.; Kurashov, V.; Golbeck, J.H.; Brudvig, G.W.; Amin, M.; Bryant, D.A. Structure of a dimeric photosystem II complex from a cyanobacterium acclimated to far-red light. J. Biol. Chem. 2023, 299, 102815. [Google Scholar] [CrossRef] [PubMed]
- Safferman, R.; Cannon, R.; Desjardins, P.; Gromov, B.; Haselkorn, R.; Sherman, L.; Shilo, M. Classification and nomenclature of viruses of cyanobacteria. Intervirology 1983, 19, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dammeyer, T.; Bagby, S.C.; Sullivan, M.B.; Chisholm, S.W.; Frankenberg-Dinkel, N. Efficient phage-mediated pigment biosynthesis in oceanic cyanobacteria. Curr. Biol. 2008, 18, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Voorhies, A.A.; Eisenlord, S.D.; Marcus, D.N.; Duhaime, M.B.; Biddanda, B.A.; Cavalcoli, J.D.; Dick, G.J. Ecological and genetic interactions between cyanobacteria and viruses in a low-oxygen mat community inferred through metagenomics and metatranscriptomics. Environ. Microbiol. 2016, 18, 358–371. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Weinheimer, A.R.; Martinez-Gutierrez, C.A.; Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 2020, 588, 141–145. [Google Scholar] [CrossRef]
- Rozenberg, A.; Oppermann, J.; Wietek, J.; Lahore, R.G.F.; Sandaa, R.-A.; Bratbak, G.; Hegemann, P.; Béjà, O. Lateral Gene Transfer of Anion-Conducting Channelrhodopsins between Green Algae and Giant Viruses. Curr. Biol. 2020, 30, 4910–4920.e4915. [Google Scholar] [CrossRef]
- Chelkha, N.; Levasseur, A.; La Scola, B.; Colson, P. Host-virus interactions and defense mechanisms for giant viruses. Ann. N. Y. Acad. Sci. 2021, 1486, 39–57. [Google Scholar] [CrossRef]
- Yoosuf, N.; Pagnier, I.; Fournous, G.; Robert, C.; La Scola, B.; Raoult, D.; Colson, P. Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae. Virus Genes 2014, 48, 218–223. [Google Scholar] [CrossRef]
- de Aquino, I.L.M.; Serafim, M.S.M.; Machado, T.B.; Azevedo, B.L.; Cunha, D.E.S.; Ullmann, L.S.; Araújo, J.P., Jr.; Abrahão, J.S. Diversity of Surface Fibril Patterns in Mimivirus Isolates. J. Virol. 2023, 97, e0182422. [Google Scholar] [CrossRef] [PubMed]
- Bisio, H.; Legendre, M.; Giry, C.; Philippe, N.; Alempic, J.-M.; Jeudy, S.; Abergel, C. Evolution of giant pandoravirus revealed by CRISPR/Cas9. Nat. Commun. 2023, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Hikida, H.; Okazaki, Y.; Zhang, R.; Nguyen, T.T.; Ogata, H. A rapid genome-wide analysis of isolated giant viruses using MinION sequencing. Environ. Microbiol. 2023, 25, 2621–2635. [Google Scholar] [CrossRef] [PubMed]
- Esmael, A.; Agarkova, I.V.; Dunigan, D.D.; Zhou, Y.; Van Etten, J.L. Viral DNA Accumulation Regulates Replication Efficiency of Chlorovirus OSy-NE5 in Two Closely Related Chlorella variabilis Strains. Viruses 2023, 15, 1341. [Google Scholar] [CrossRef]
- Fernández-García, J.L.; de Ory, A.; Brussaard, C.P.D.; de Vega, M. Phaeocystis globosa Virus DNA Polymerase X: A “Swiss Army knife”, Multifunctional DNA polymerase-lyase-ligase for Base Excision Repair. Sci. Rep. 2017, 7, 6907. [Google Scholar] [CrossRef]
- Derelle, E.; Monier, A.; Cooke, R.; Worden, A.Z.; Grimsley, N.H.; Moreau, H. Diversity of Viruses Infecting the Green Microalga Ostreococcus lucimarinus. J. Virol. 2015, 89, 5812–5821. [Google Scholar] [CrossRef]
- Delaroque, N.; Maier, I.; Knippers, R.; Müller, D.G. Persistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J. Gen. Virol. 1999, 80 Pt 6, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef]
- Kehr, J.C.; Dittmann, E. Biosynthesis and function of extracellular glycans in cyanobacteria. Life 2015, 5, 164–180. [Google Scholar] [CrossRef]
- Walhout, A.J.; Temple, G.; Brasch, M.; Hartley, J.; Lorson, M.; Van Den Heuvel, S.; Vidal, M. GATEWAY recombinational cloning: Application to the cloning of large numbers of open reading frames or ORFeomes. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2000; Volume 328, pp. 575–592, IN7. [Google Scholar]
- Hartley, J.L.; Temple, G.F.; Brasch, M.A. DNA cloning using in vitro site-specific recombination. Genome Res. 2000, 10, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Dreze, M.; Monachello, D.; Lurin, C.; Cusick, M.E.; Hill, D.E.; Vidal, M.; Braun, P. High-Quality Binary Interactome Mapping. In Methods in Enzymology; Academic Press: New York, NY, USA, 2010; Volume 470, pp. 281–315. [Google Scholar]
- Sánchez-Baracaldo, P. Origin of marine planktonic cyanobacteria. Sci. Rep. 2015, 5, 17418. [Google Scholar] [CrossRef]
- Ulloa, O.; Henríquez-Castillo, C.; Ramírez-Flandes, S.; Plominsky, A.M.; Murillo, A.A.; Morgan-Lang, C.; Hallam, S.J.; Stepanauskas, R. The cyanobacterium Prochlorococcus has divergent light-harvesting antennae and may have evolved in a low-oxygen ocean. Proc. Natl. Acad. Sci. USA 2021, 118, e2025638118. [Google Scholar] [CrossRef] [PubMed]
- El-Seedi, H.R.; El-Mallah, M.F.; Yosri, N.; Alajlani, M.; Zhao, C.; Mehmood, M.A.; Du, M.; Ullah, H.; Daglia, M.; Guo, Z.; et al. Review of Marine Cyanobacteria and the Aspects Related to Their Roles: Chemical, Biological Properties, Nitrogen Fixation and Climate Change. Mar. Drugs 2023, 21, 439. [Google Scholar] [CrossRef]
- Flombaum, P.; Gallegos, J.L.; Gordillo, R.A.; Rincón, J.; Zabala, L.L.; Jiao, N.; Karl, D.M.; Li, W.K.; Lomas, M.W.; Veneziano, D.; et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 9824–9829. [Google Scholar] [CrossRef]
- Puxty, R.J.; Millard, A.D.; Evans, D.J.; Scanlan, D.J. Shedding new light on viral photosynthesis. Photosynth. Res. 2015, 126, 71–97. [Google Scholar] [CrossRef]
- James, J.E.; Nelson, P.G.; Masel, J. Differential Retention of Pfam Domains Contributes to Long-term Evolutionary Trends. Mol. Biol. Evol. 2023, 40, msad073. [Google Scholar] [CrossRef] [PubMed]
- Godbold, J.A.; Calosi, P. Ocean acidification and climate change: Advances in ecology and evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120448. [Google Scholar] [CrossRef] [PubMed]
- Beardall, J.; Stojkovic, S.; Larsen, S. Living in a high CO2 world: Impacts of global climate change on marine phytoplankton. Plant Ecol. Divers. 2009, 2, 191–205. [Google Scholar] [CrossRef]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: A formidable predictive challenge 1. J. Phycol. 2010, 46, 220–235. [Google Scholar] [CrossRef]
- Shestakov, S.V.; Karbysheva, E.A. The role of viruses in the evolution of cyanobacteria. Biol. Bull. Rev. 2015, 5, 527–537. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Doolittle, W.F.; Papke, R.T.; Gogarten, J.P. Intertwined evolutionary histories of marine Synechococcus and Prochlorococcus marinus. Genome Biol. Evol. 2009, 1, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, G.; Bielawski, J.P.; Shmoish, M.; Scanlan, D.J.; Sabehi, G.; Béjà, O. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ. Microbiol. 2005, 7, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Jaffe, J.D.; Coleman, M.L.; Futschik, M.E.; Axmann, I.M.; Rector, T.; Kettler, G.; Sullivan, M.B.; Steen, R.; Hess, W.R.; et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 2007, 449, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Shen, Y.-P.; Ma, L.-G.; Pan, Y.; Du, Y.-L.; Wang, D.-H.; Yang, J.-Y.; Hu, L.-D.; Liu, X.-F.; Dong, C.-X.; et al. Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol. 2004, 135, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.E.; Breton, G.; Pruneda-Paz, J.L. Construction of Arabidopsis Transcription Factor ORFeome Collections and Identification of Protein-DNA Interactions by High-Throughput Yeast One-Hybrid Screens. Methods Mol. Biol. 2018, 1794, 151–182. [Google Scholar]
- Lamesch, P.; Li, N.; Milstein, S.; Fan, C.; Hao, T.; Szabo, G.; Hu, Z.; Venkatesan, K.; Bethel, G.; Martin, P.; et al. hORFeome v3.1: A resource of human open reading frames representing over 10,000 human genes. Genomics 2007, 89, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rual, J.-F.; Hill, D.E.; Vidal, M. ORFeome projects: Gateway between genomics and omics. Curr. Opin. Chem. Biol. 2004, 8, 20–25. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Yamamoto, N.; Zweifel, A.E.; Nakamichi, T.; Huang, H.-K.; Mendez-Rios, J.D.; Franca-Koh, J.; Boorgula, M.P.; Fujita, K.; Suzuki, K.-I.; et al. The Escherichia coli K-12 ORFeome: A resource for comparative molecular microbiology. BMC Genom. 2010, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Özkan, E.; Carrillo, R.A.; Eastman, C.L.; Weiszmann, R.; Waghray, D.; Johnson, K.G.; Zinn, K.; Celniker, S.E.; Garcia, K.C. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell 2013, 154, 228–239. [Google Scholar] [CrossRef]
- Ghamsari, L.; Balaji, S.; Shen, Y.; Yang, X.; Balcha, D.; Fan, C.; Hao, T.; Yu, H.; Papin, J.A.; Salehi-Ashtiani, K. Genome-wide functional annotation and structural verification of metabolic ORFeome of Chlamydomonas reinhardtii. BMC Genom. 2011, 12 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Pellet, J.; Tafforeau, L.; Lucas-Hourani, M.; Navratil, V.; Meyniel, L.; Achaz, G.; Guironnet-Paquet, A.; Aublin-Gex, A.; Caignard, G.; Cassonnet, P.; et al. ViralORFeome: An integrated database to generate a versatile collection of viral ORFs. Nucleic Acids Res. 2010, 38, D371–D378. [Google Scholar] [CrossRef]
- Fu, W.; Nelson, D.R.; Mystikou, A.; Daakour, S.; Salehi-Ashtiani, K. Advances in microalgal research and engineering development. Curr. Opin. Biotechnol. 2019, 59, 157–164. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daakour, S.; Nelson, D.R.; Fu, W.; Jaiswal, A.; Dohai, B.; Alzahmi, A.S.; Koussa, J.; Huang, X.; Shen, Y.; Twizere, J.-C.; et al. Adaptive Evolution Signatures in Prochlorococcus: Open Reading Frame (ORF)eome Resources and Insights from Comparative Genomics. Microorganisms 2024, 12, 1720. https://doi.org/10.3390/microorganisms12081720
Daakour S, Nelson DR, Fu W, Jaiswal A, Dohai B, Alzahmi AS, Koussa J, Huang X, Shen Y, Twizere J-C, et al. Adaptive Evolution Signatures in Prochlorococcus: Open Reading Frame (ORF)eome Resources and Insights from Comparative Genomics. Microorganisms. 2024; 12(8):1720. https://doi.org/10.3390/microorganisms12081720
Chicago/Turabian StyleDaakour, Sarah, David R. Nelson, Weiqi Fu, Ashish Jaiswal, Bushra Dohai, Amnah Salem Alzahmi, Joseph Koussa, Xiaoluo Huang, Yue Shen, Jean-Claude Twizere, and et al. 2024. "Adaptive Evolution Signatures in Prochlorococcus: Open Reading Frame (ORF)eome Resources and Insights from Comparative Genomics" Microorganisms 12, no. 8: 1720. https://doi.org/10.3390/microorganisms12081720
APA StyleDaakour, S., Nelson, D. R., Fu, W., Jaiswal, A., Dohai, B., Alzahmi, A. S., Koussa, J., Huang, X., Shen, Y., Twizere, J.-C., & Salehi-Ashtiani, K. (2024). Adaptive Evolution Signatures in Prochlorococcus: Open Reading Frame (ORF)eome Resources and Insights from Comparative Genomics. Microorganisms, 12(8), 1720. https://doi.org/10.3390/microorganisms12081720