
Molecular Evolutionary Analyses of Shiga toxin type 2 subunit A Gene in the Enterohemorrhagic Escherichia coli (EHEC)


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains Used in This Study
2.2. Time-Scaled Phylogenetic Analyses
2.3. Phylogenetic Distance Analyses
2.4. Phylodynamic Analyses
2.5. Selective Pressure Analyses
2.6. Construction of the 3D Structure of Stx2A
2.7. Conformational B-Cell Epitope Prediction
3. Results
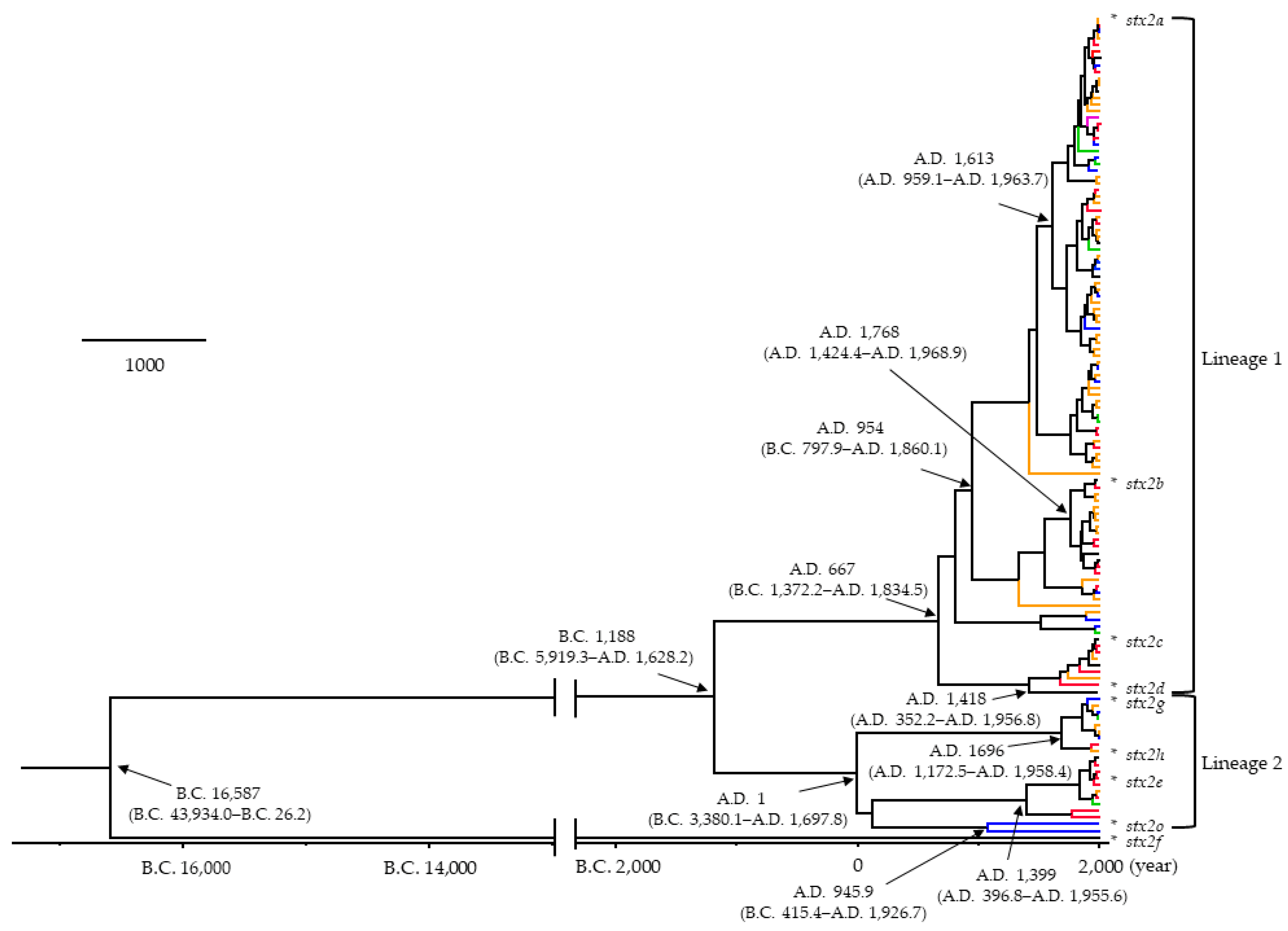
3.1. Time-Scaled Phylogeny
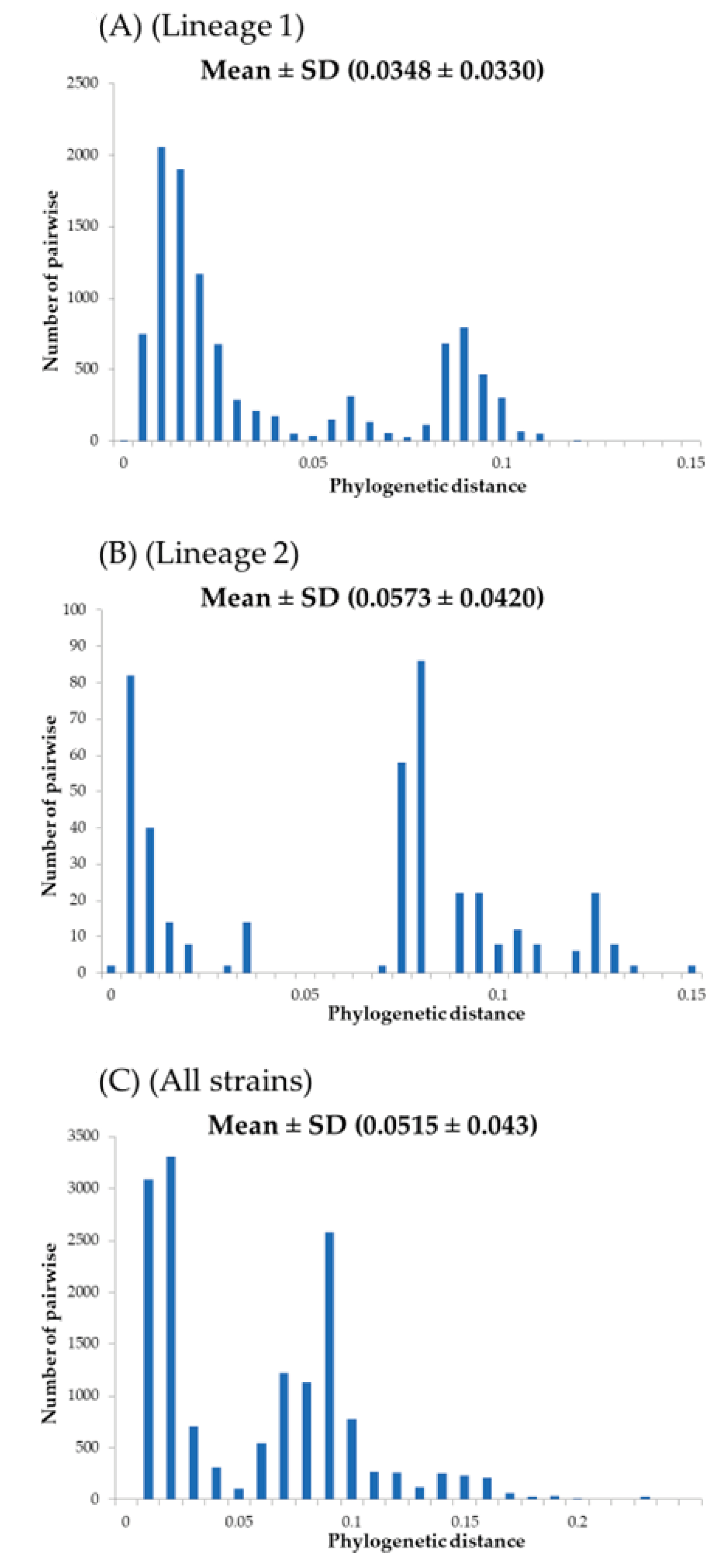
3.2. Phylogenetic Distances
3.3. Phylodynamics
3.4. Relationships between Selective Pressure Sites and Active Sites
3.5. Conformational Epitope Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Donnenberg, M.S.; Whittam, T.S. Pathogenesis and evolution of virulence in enteropathogenic and enterohemorrhagic Escherichia coli. J. Clin. Investig. 2001, 107, 539–548. [Google Scholar] [CrossRef]
- Mueller, M.; Tainter, C.R. Escherichia coli infection. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Scheiring, J.; Andreoli, S.P.; Zimmerhackl, L.B. Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS). Pediatr. Nephrol. 2008, 23, 1749–1760. [Google Scholar] [CrossRef]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar] [CrossRef] [PubMed]
- de Sablet, T.; Bertin, Y.; Vareille, M.; Girardeau, J.-P.; Garrivier, A.; Gobert, A.P.; Martin, C. Differential expression of stx2 variants in Shiga toxin-producing Escherichia coli belonging to seropathotypes A and C. Microbiology 2008, 154, 176–186. [Google Scholar] [CrossRef]
- Ogura, Y.; Mondal, S.I.; Islam, M.R.; Mako, T.; Arisawa, K.; Katsura, K.; Ooka, T.; Gotoh, Y.; Murase, K.; Ohnishi, M. The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157: H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 2015, 5, 16663. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Sato, T.; Kawakami, S.; Ohta, T.; Noda, M.; Hamabata, T. Receptor affinity, stability and binding mode of Shiga toxins are determinants of toxicity. Microb. Pathog. 2007, 43, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Tumer, N.E. Do the A subunits contribute to the differences in the toxicity of Shiga toxin 1 and Shiga toxin 2? Toxins 2015, 7, 1467–1485. [Google Scholar] [CrossRef]
- Lee, J.E.; Reed, J.; Shields, M.S.; Spiegel, K.M.; Farrell, L.D.; Sheridan, P.P. Phylogenetic analysis of Shiga toxin 1 and Shiga toxin 2 genes associated with disease outbreaks. BMC Microbiol. 2007, 7, 109. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Awosika, J.; Baldwin, C.; Bishop-Lilly, K.A.; Biswas, B.; Broomall, S.; Chain, P.S.; Chertkov, O.; Chokoshvili, O.; Coyne, S. Genomic comparison of Escherichia coli O104: H4 isolates from 2009 and 2011 reveals plasmid, and prophage heterogeneity, including shiga toxin encoding phage stx2. PLoS ONE 2012, 7, e48228. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2, EHEC-0024. [Google Scholar] [CrossRef]
- Johannes, L.; Römer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Acheson, D.W.; Waldor, M.K. Isogenic lysogens of diverse Shiga toxin 2-encoding bacteriophages produce markedly different amounts of Shiga toxin. Infect. Immun. 1999, 67, 6710–6714. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Matsui, T.; Takita, E.; Kadoyama, Y.; Makino, S.-I.; Kato, K.; Sawada, K.; Hamabata, T. Evaluation of recombinant forms of the shiga toxin variant Stx2eB subunit and non-toxic mutant Stx2e as vaccine candidates against porcine edema disease. J. Vet. Med. Sci. 2013, 75, 1309–1315. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157: H7 and of Shiga toxin on eukaryotic ribosomes: RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef]
- Saxena, S.K.; O’brien, A.; Ackerman, E. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28 S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 1989, 264, 596–601. [Google Scholar] [CrossRef]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr. Res. 2001, 50, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Szymczak-Kulus, K.; Weidler, S.; Bereznicka, A.; Mikolajczyk, K.; Kaczmarek, R.; Bednarz, B.; Zhang, T.; Urbaniak, A.; Olczak, M.; Park, E.Y. Human Gb3/CD77 synthase produces P1 glycotope-capped N-glycans, which mediate Shiga toxin 1 but not Shiga toxin 2 cell entry. J. Biol. Chem. 2021, 296, 100299. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.S.; Lemey, P.; Suchard, M.A.; Rambaut, A.; Baele, G. Online Bayesian phylodynamic inference in BEAST with application to epidemic reconstruction. Mol. Biol. Evol. 2020, 37, 1832–1842. [Google Scholar] [CrossRef]
- Shakya, M.; Ahmed, S.A.; Davenport, K.W.; Flynn, M.C.; Lo, C.-C.; Chain, P.S. Standardized phylogenetic and molecular evolutionary analysis applied to species across the microbial tree of life. Sci. Rep. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Yu, F.; Cienfuegos-Gallet, A.V.; Cunningham, M.H.; Jin, Y.; Wang, B.; Kreiswirth, B.N.; Chen, L. Molecular evolution and adaptation of livestock-associated methicillin-resistant Staphylococcus aureus (LA-MRSA) sequence type 9. Msystems 2021, 6, e0049221. [Google Scholar] [CrossRef]
- Gill, A.; Dussault, F.; McMahon, T.; Petronella, N.; Wang, X.; Cebelinski, E.; Scheutz, F.; Weedmark, K.; Blais, B.; Carrillo, C. Characterization of Atypical Shiga Toxin Gene Sequences and Description of Stx2j, a New Subtype. J. Clin. Microbiol. 2022, 60, e0222921. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software ’EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Fourment, M.; Gibbs, M.J. PATRISTIC: A program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 2006, 6, 1. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2021, 2199, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 2005, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, Z.; Zhang, L.; Yan, D.; Mao, T.; Tang, K.; Qiu, T.; Cao, Z. SEPPA 3.0-enhanced spatial epitope prediction enabling glycoprotein antigens. Nucleic Acids Res. 2019, 47, W388–W394. [Google Scholar] [CrossRef] [PubMed]
- Yano, B.; Taniguchi, I.; Gotoh, Y.; Hayashi, T.; Nakamura, K. Dynamic changes in Shiga toxin (Stx) 1 transducing phage throughout the evolution of O26:H11 Stx-producing Escherichia coli. Sci. Rep. 2023, 13, 4935. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.S.; Rix, G.; Mengiste, A.A.; Álvarez, B.; Seo, D.; Chen, H.; Hurtado, J.E.; Zhang, Q.; García-García, J.D.; Heins, Z.J. In vivo hypermutation and continuous evolution. Nat. Rev. Methods Primers 2022, 2, 36. [Google Scholar] [CrossRef]
- Bergeron, L.A.; Besenbacher, S.; Zheng, J.; Li, P.; Bertelsen, M.F.; Quintard, B.; Hoffman, J.I.; Li, Z.; St. Leger, J.; Shao, C.; et al. Evolution of the germline mutation rate across vertebrates. Nature 2023, 615, 285–291. [Google Scholar] [CrossRef]
- Duchêne, S.; Holt, K.E.; Weill, F.-X.; Le Hello, S.; Hawkey, J.; Edwards, D.J.; Fourment, M.; Holmes, E.C. Genome-scale rates of evolutionary change in bacteria. Microb. Genom. 2016, 2, e000094. [Google Scholar] [CrossRef]
- Parag, K.V.; du Plessis, L.; Pybus, O.G. Jointly Inferring the Dynamics of Population Size and Sampling Intensity from Molecular Sequences. Mol. Biol. Evol. 2020, 37, 2414–2429. [Google Scholar] [CrossRef]
- Karcher, M.D.; Carvalho, L.M.; Suchard, M.A.; Dudas, G.; Minin, V.N. Estimating effective population size changes from preferentially sampled genetic sequences. PLOS Comput. Biol. 2020, 16, e1007774. [Google Scholar] [CrossRef]
- Volz, E.M.; Didelot, X. Modeling the growth and decline of pathogen effective population size provides insight into epidemic dynamics and drivers of antimicrobial resistance. Syst. Biol. 2018, 67, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.; Griffith, K.H.B.; David, P. Anderson. Managing the Beef Cattle Herd through the Cattle Cycle. Surviv. Farm Econ. Downturn 2017, 1, 54–58. [Google Scholar]
- Bielaszewska, M.; Friedrich, A.W.; Aldick, T.; Schurk-Bulgrin, R.; Karch, H. Shiga toxin activatable by intestinal mucus in Escherichia coli isolated from humans: Predictor for a severe clinical outcome. Clin. Infect. Dis. 2006, 43, 1160–1167. [Google Scholar] [CrossRef]
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat. Immunol. 2002, 3, 1033–1040. [Google Scholar] [CrossRef]
- Brunham, R.C.; Plummer, F.A.; Stephens, R.S. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infect. Immun. 1993, 61, 2273–2276. [Google Scholar] [CrossRef]
- Shirai, T.; Akagawa, M.; Makino, M.; Ishii, M.; Arai, A.; Nagasawa, N.; Sada, M.; Kimura, R.; Okayama, K.; Ishioka, T. Molecular Evolutionary Analyses of the Pseudomonas-Derived Cephalosporinase Gene. Microorganisms 2023, 11, 635. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30. [Google Scholar] [CrossRef]
- Smith, M.J.; Melton-Celsa, A.R.; Sinclair, J.F.; Carvalho, H.M.; Robinson, C.M.; O’Brien, A.D. Monoclonal antibody 11E10, which neutralizes shiga toxin type 2 (Stx2), recognizes three regions on the Stx2 A subunit, blocks the enzymatic action of the toxin in vitro, and alters the overall cellular distribution of the toxin. Infect. Immun. 2009, 77, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Pakbin, B.; Bruck, W.M.; Rossen, J.W.A. Virulence Factors of Enteric Pathogenic Escherichia coli: A Review. Int. J. Mol. Sci. 2021, 22, 9922. [Google Scholar] [CrossRef]
- Mullaney, B.; Pallavicini, M.; Marks, J. Epitope mapping of neutralizing botulinum neurotoxin A antibodies by phage display. Infect. Immun. 2001, 69, 6511–6514. [Google Scholar] [CrossRef]
- Hijnen, M.; Mooi, F.R.; van Gageldonk, P.G.; Hoogerhout, P.; King, A.J.; Berbers, G.A. Epitope structure of the Bordetella pertussis protein P. 69 pertactin, a major vaccine component and protective antigen. Infect. Immun. 2004, 72, 3716–3723. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substitution Model | Gamma Shape | Proportion Invariant | Clock Models | Demographic Models | Chain Length | Log Every | |
|---|---|---|---|---|---|---|---|
| All strains (125 strain) | HKY-Γ-I | 0.398 | 0.304 | Relaxed Clock Exponential | Coalescent Constant Population | 250,000,000 | 10,000 |
| All strains (124 strains except stx2f) | HKY-Γ-I | 0.398 | 0.304 | Relaxed Clock Exponential | Coalescent Bayesian Skyline | 250,000,000 | 10,000 |
| Lineage 1 (103 strains) | TPM2uf-Γ-I | 0.741 | 0.724 | Relaxed Clock Exponential | Coalescent Bayesian Skyline | 250,000,000 | 10,000 |
| Lineage 2 (21 strains) | HKY-Γ | 0.104 | - | Relaxed Clock Exponential | Coalescent Bayesian Skyline | 250,000,000 | 10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, R.; Kimura, H.; Shirai, T.; Hayashi, Y.; Sato-Fujimoto, Y.; Kamitani, W.; Ryo, A.; Tomita, H. Molecular Evolutionary Analyses of Shiga toxin type 2 subunit A Gene in the Enterohemorrhagic Escherichia coli (EHEC). Microorganisms 2024, 12, 1812. https://doi.org/10.3390/microorganisms12091812
Kimura R, Kimura H, Shirai T, Hayashi Y, Sato-Fujimoto Y, Kamitani W, Ryo A, Tomita H. Molecular Evolutionary Analyses of Shiga toxin type 2 subunit A Gene in the Enterohemorrhagic Escherichia coli (EHEC). Microorganisms. 2024; 12(9):1812. https://doi.org/10.3390/microorganisms12091812
Chicago/Turabian StyleKimura, Ryusuke, Hirokazu Kimura, Tatsuya Shirai, Yuriko Hayashi, Yuka Sato-Fujimoto, Wataru Kamitani, Akihide Ryo, and Haruyoshi Tomita. 2024. "Molecular Evolutionary Analyses of Shiga toxin type 2 subunit A Gene in the Enterohemorrhagic Escherichia coli (EHEC)" Microorganisms 12, no. 9: 1812. https://doi.org/10.3390/microorganisms12091812