

A Genome-Focused Investigation Reveals the Emergence of a Mycobacterium tuberculosis Strain Related to Multidrug-Resistant Tuberculosis in the Amazon Region of Brazil

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Population and Epidemiological Information
2.3. Culture, Drug Susceptibility and Molecular Tests
2.4. Whole-Genome Sequencing
2.5. Bioinformatic Analysis of Genomic Data
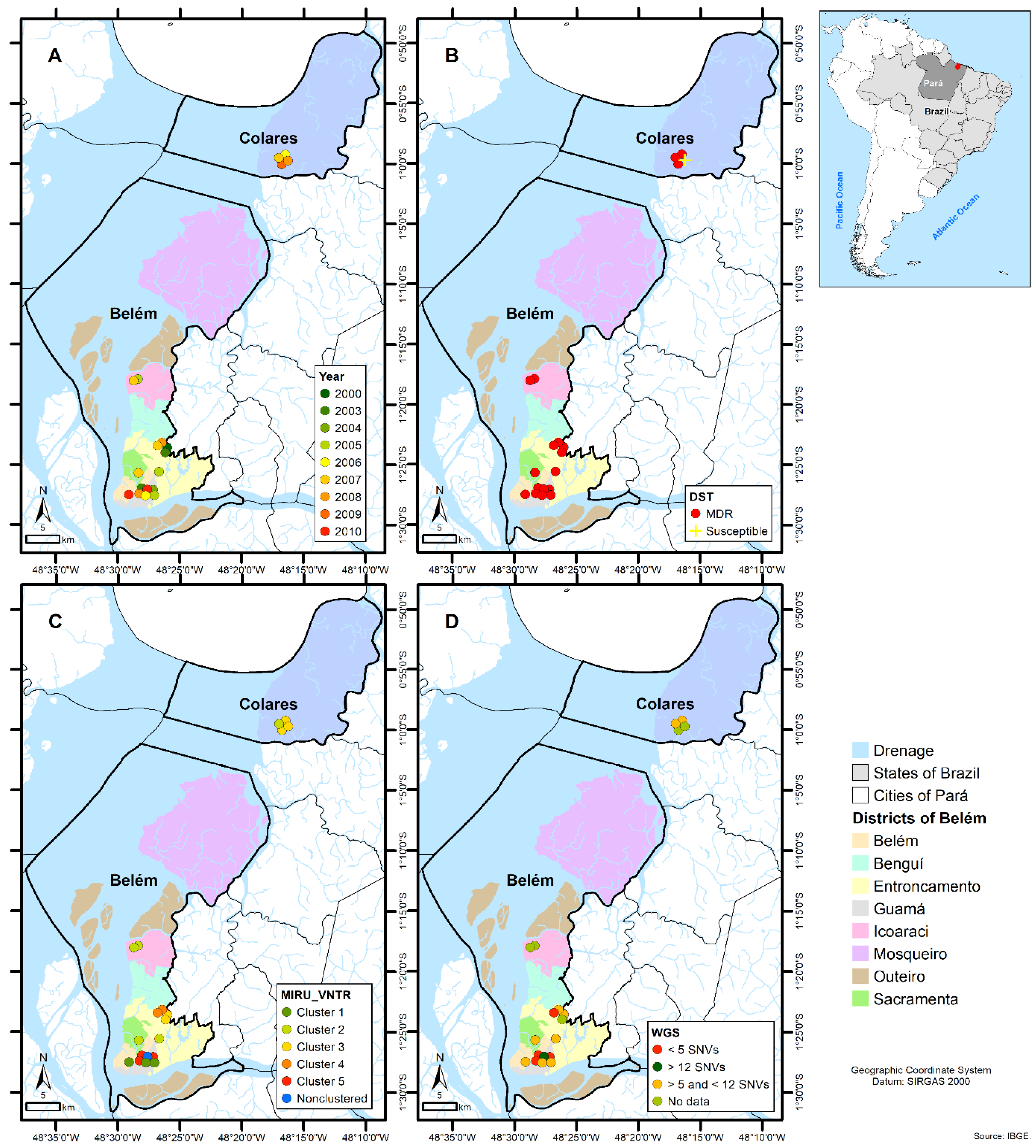
2.6. Geographic Information System
2.7. Transmission Analysis
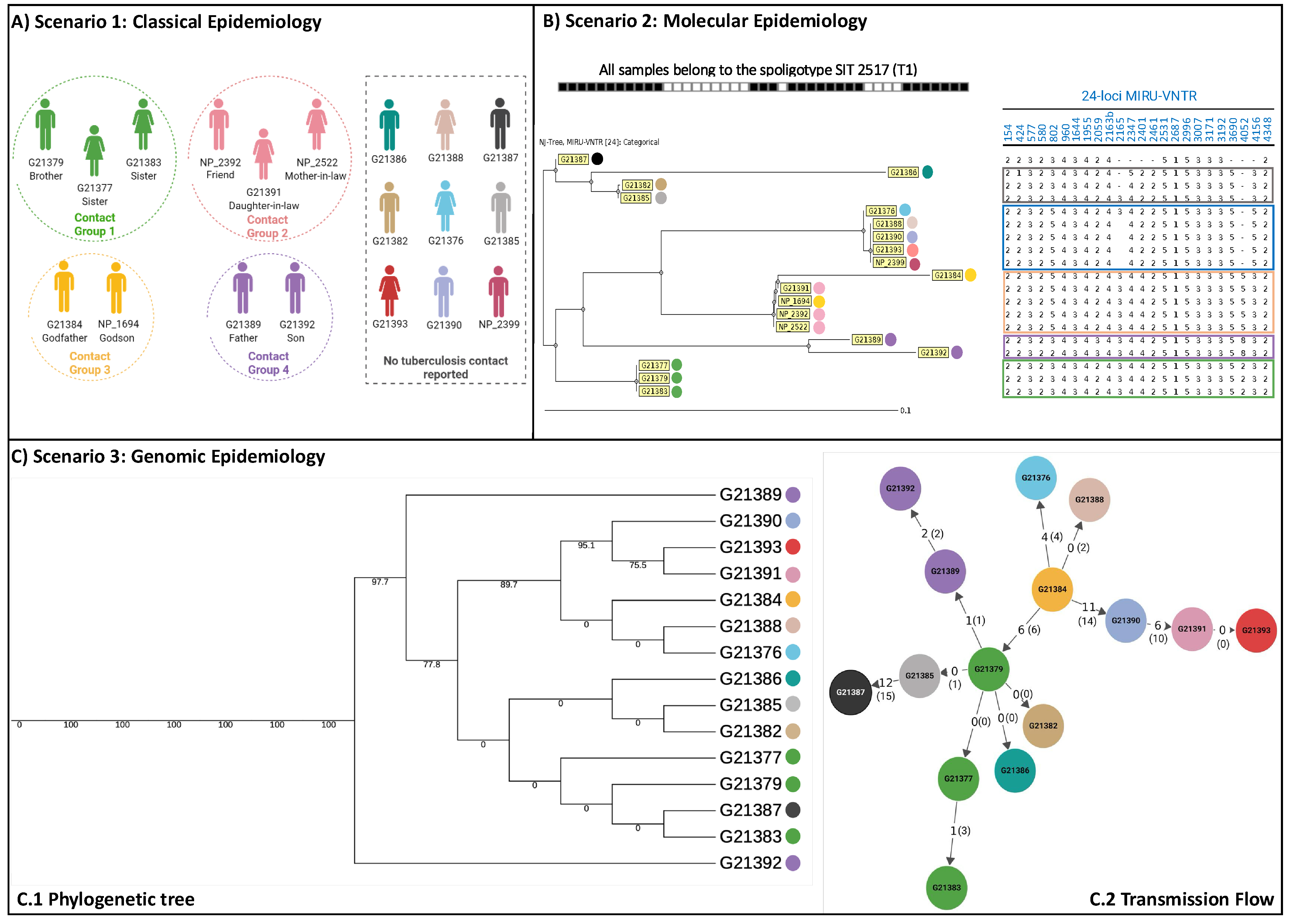
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, L.L.; Yu, M.A.; Sacks, J.A.; Barnadas, C.; Pereyaslov, D.; Cognat, S.; Briand, S.; Ryan, M.J.; Samaan, G. Global Genomic Surveillance Strategy for Pathogens with Pandemic and Epidemic Potential 2022–2032. Bull. World Health Organ. 2022, 100, 239. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.D.; Raangs, E.C.; Rosema, S.; Veloo, A.C.M.; et al. Application of next Generation Sequencing in Clinical Microbiology and Infection Prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Alleweldt, F.; Kara, Ş.; Best, K.; Aarestrup, F.M.; Beer, M.; Bestebroer, T.M.; Campos, J.; Casadei, G.; Chinen, I.; Van Domselaar, G.; et al. Economic Evaluation of Whole Genome Sequencing for Pathogen Identification and Surveillance—Results of Case Studies in Europe and the Americas 2016 to 2019. Eurosurveillance 2021, 26, 1900606. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2023; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Kargarpour Kamakoli, M.; Hadifar, S.; Khanipour, S.; Farmanfarmaei, G.; Fateh, A.; Siadat, S.D.; Vaziri, F. Comparison of MIRU-VNTR Genotyping between Old and Fresh Clinical Samples in Tuberculosis. Infect. Dis. Lond. Engl. 2019, 51, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Black, A.T.; Hamblion, E.L.; Buttivant, H.; Anderson, S.R.; Stone, M.; Casali, N.; Drobniewski, F.; Nwoguh, F.; Marshall, B.G.; Booth, L. Tracking and Responding to an Outbreak of Tuberculosis Using MIRU-VNTR Genotyping and Whole Genome Sequencing as Epidemiological Tools. J. Public Health 2018, 40, e66–e73. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Merker, M.; Kohl, T.A.; Crook, D.W.; Niemann, S.; Peto, T.E.A. Whole Genome Sequencing for M/XDR Tuberculosis Surveillance and for Resistance Testing. Clin. Microbiol. Infect. 2017, 23, 161–166. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the Biology of Mycobacterium tuberculosis from the Complete Genome Sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Siddiqi, S.; Ahmed, A.; Asif, S.; Behera, D.; Javaid, M.; Jani, J.; Jyoti, A.; Mahatre, R.; Mahto, D.; Richter, E.; et al. Direct Drug Susceptibility Testing of Mycobacterium tuberculosis for Rapid Detection of Multidrug Resistance Using the Bactec MGIT 960 System: A Multicenter Study. J. Clin. Microbiol. 2012, 50, 435–440. [Google Scholar] [CrossRef]
- Nelson, K.N.; Talarico, S.; Poonja, S.; McDaniel, C.J.; Cilnis, M.; Chang, A.H.; Raz, K.; Noboa, W.S.; Cowan, L.; Shaw, T.; et al. Mutation of Mycobacterium tuberculosis and Implications for Using Whole-Genome Sequencing for Investigating Recent Tuberculosis Transmission. Front. Public Health 2021, 9, 790544. [Google Scholar] [CrossRef]
- Jiang, Q.; Liu, H.-C.; Liu, Q.-Y.; Phelan, J.E.; Tao, F.-X.; Zhao, X.-Q.; Wang, J.; Glynn, J.R.; Takiff, H.E.; Clark, T.G.; et al. The Evolution and Transmission Dynamics of Multidrug-Resistant Tuberculosis in an Isolated High-Plateau Population of Tibet, China. Microbiol. Spectr. 2023, 11, e03991-22. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, Z.; Tian, D.; Zhou, M.; Hu, Y.; Wu, J.; Wang, T.; Li, Y.; Chen, J. Transmission of Multidrug-Resistant Mycobacterium tuberculosis in Wuhan, China: A Retrospective Molecular Epidemiological Study. Medicine 2022, 101, e28751. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.J.; Schaaf, H.S.; Mandalakas, A.; Chiappini, E.; Zumla, A.; Marais, B.J. Preventing the Spread of Multidrug-Resistant Tuberculosis and Protecting Contacts of Infectious Cases. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2017, 23, 147–153. [Google Scholar] [CrossRef]
- Salvatore, P.P.; Kendall, E.A.; Seabrook, D.; Brown, J.; Durham, G.H.; Dowdy, D.W. Projecting the Impact of Variable MDR-TB Transmission Efficiency on Long-Term Epidemic Trends in South Africa and Vietnam. Sci. Rep. 2019, 9, 18099. [Google Scholar] [CrossRef]
- Viney, K.; Linh, N.N.; Gegia, M.; Zignol, M.; Glaziou, P.; Ismail, N.; Kasaeva, T.; Mirzayev, F. New Definitions of Pre-Extensively and Extensively Drug-Resistant Tuberculosis: Update from the World Health Organization. Eur. Respir. J. 2021, 57, 2100361. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Announces Updated Definitions of Extensively Drug-Resistant Tuberculosis; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Conceição, E.C.E.C.; Rastogi, N.; Couvin, D.; Lopes, M.L.M.L.; Furlaneto, I.P.I.P.; Gomes, H.M.H.M.; Vasconcellos, S.E.G.S.E.G.; Suffys, P.N.P.N.; Schneider, M.P.C.M.P.C.; de Sousa, M.S.M.S.; et al. Genetic Diversity of Mycobacterium tuberculosis from Pará, Brazil, Reveals a Higher Frequency of Ancestral Strains than Previously Reported in South America. Infect. Genet. Evol. 2017, 56, 62–74. [Google Scholar] [CrossRef]
- Gomes, H.M.; Elias, A.R.; Oelemann, M.A.C.; Pereira, M.A.D.S.; Montes, F.F.O.; Marsico, A.G.; Kritski, A.L.; Filho, L.D.A.; Caldas, P.C.; Possuelo, L.G.; et al. Spoligotypes of Mycobacterium tuberculosis Complex Isolates from Patients Residents of 11 States of Brazil. Infect. Genet. Evol. 2012, 12, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Hadi, S.A.; Kolte, I.V.; Brenner, E.P.; Cunha, E.A.T.; Simonsen, V.; Ferrazoli, L.; Villela, D.A.M.; Santos, R.S.; Ravi, J.; Sreevatsan, S.; et al. Identification of a Predominant Genotype of Mycobacterium tuberculosis in Brazilian Indigenous Population. Sci. Rep. 2021, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- Conceição, E.C.; da Conceição, M.L.; Marcon, D.J.; Loubser, J.; Andrade, G.L.; Silva, S.P.d.; Cruz, A.C.R.; Sharma, A.; Suffys, P.; Lima, K.V.B. Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil. Microorganisms 2023, 11, 132. [Google Scholar] [CrossRef]
- Walter, K.S.; Dos Santos, P.C.P.; Gonçalves, T.O.; da Silva, B.O.; da Silva Santos, A.; de Cássia Leite, A.; da Silva, A.M.; Figueira Moreira, F.M.; de Oliveira, R.D.; Lemos, E.F.; et al. The Role of Prisons in Disseminating Tuberculosis in Brazil: A Genomic Epidemiology Study. Lancet Reg. Health Am. 2022, 9, 100186. [Google Scholar] [CrossRef]
- Conceição, E.C.; Salvato, R.S.; Gomes, K.M.; Guimarães, A.E.D.S.; da Conceição, M.L.; Souza EGuimarães, R.J.P.; Sharma, A.; Furlaneto, I.P.; Barcellos, R.B.; Bollela, V.R.; et al. Molecular Epidemiology of Mycobacterium tuberculosis in Brazil before the Whole Genome Sequencing Era: A Literature Review. Mem. Inst. Oswaldo Cruz 2021, 116, e200517. [Google Scholar] [CrossRef] [PubMed]
- Monir, B.B.; Sultana, S.S.; Tarafder, S. 24 Loci MIRU-VNTR Analysis and Pattern of Drug Resistance in Pre-Extensively Drug Resistant Pulmonary Tuberculosis in Bangladesh. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2022, 102, 105304. [Google Scholar] [CrossRef]
- Wyllie, D.H.; Davidson, J.A.; Grace Smith, E.; Rathod, P.; Crook, D.W.; Peto, T.E.A.; Robinson, E.; Walker, T.; Campbell, C. A Quantitative Evaluation of MIRU-VNTR Typing Against Whole-Genome Sequencing for Identifying Mycobacterium tuberculosis Transmission: A Prospective Observational Cohort Study. EBioMedicine 2018, 34, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Dale, K.; Globan, M.; Horan, K.; Sherry, N.; Ballard, S.; Tay, E.L.; Bittmann, S.; Meagher, N.; Price, D.J.; Howden, B.P.; et al. Whole Genome Sequencing for Tuberculosis in Victoria, Australia: A Genomic Implementation Study from 2017 to 2020. Lancet Reg. Health-West. Pac. 2022, 28, 100556. [Google Scholar] [CrossRef] [PubMed]
- Supply, P. Multilocus Variable Number Tandem Repeat Genotyping of Mycobacterium tuberculosis. Tech. Guide 2005, 6, 1–74. [Google Scholar]
- Weniger, T.; Krawczyk, J.; Supply, P.; Niemann, S.; Harmsen, D. MIRU-VNTRplus: A Web Tool for Polyphasic Genotyping of Mycobacterium tuberculosis Complex Bacteria. Nucleic Acids Res. 2010, 38, W326–W331. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinform. Oxf. Engl. 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Verza, M.; Scheffer, M.C.; Salvato, R.S.; Schorner, M.A.; Barazzetti, F.H.; de Melo Machado, H.; Medeiros, T.F.; Rovaris, D.B.; Portugal, I.; Viveiros, M.; et al. Genomic Epidemiology of Mycobacterium tuberculosis in Santa Catarina, Southern Brazil. Sci. Rep. 2020, 10, 12891. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, C.S.; Namburete, E.I.; Rodrigues Plaça, J.; Peronni, K.; Dippenaar, A.; Warren, R.M.; Silva, W.A.; Bollela, V.R. Accuracy of Whole Genome Sequencing versus Phenotypic (MGIT) and Commercial Molecular Tests for Detection of Drug-Resistant Mycobacterium tuberculosis Isolated from Patients in Brazil and Mozambique. Tuberculosis 2018, 10, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Brynildsrud, O.B.; Pepperell, C.S.; Suffys, P.; Grandjean, L.; Monteserin, J.; Debech, N.; Bohlin, J.; Alfsnes, K.; Pettersson, J.O.H.; Kirkeleite, I.; et al. Global Expansion of Mycobacterium tuberculosis Lineage 4 Shaped by Colonial Migration and Local Adaptation. Sci. Adv. 2018, 4, eaat5869. [Google Scholar] [CrossRef] [PubMed]
- Salvato, R.S.; Reis, A.J.; Schiefelbein, S.H.; Gómez, M.A.A.; Salvato, S.S.; da Silva, L.V.; Costa, E.R.D.; Unis, G.; Dias, C.F.; Viveiros, M.; et al. Genomic-Based Surveillance Reveals High Ongoing Transmission of Multi-Drug-Resistant Mycobacterium tuberculosis in Southern Brazil. Int. J. Antimicrob. Agents 2021, 58, 106401. [Google Scholar] [CrossRef] [PubMed]
- Colangeli, R.; Gupta, A.; Vinhas, S.A.; Venkata, U.D.C.; Kim, S.; Grady, C.; Jones-López, E.C.; Soteropoulos, P.; Palaci, M.; Marques-Rodrigues, P.; et al. Mycobacterium tuberculosis Progresses through Two Phases of Latent Infection in Humans. Nat. Commun. 2020, 11, 4870. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.E.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Oppong, Y.E.A.; Campino, S.; O’Grady, J.; McNerney, R.; Hibberd, M.L.; Viveiros, M.; et al. Integrating Informatics Tools and Portable Sequencing Technology for Rapid Detection of Resistance to Anti-Tuberculous Drugs. Genome Med. 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- WHO. Catalogue of Mutations in Mycobacterium tuberculosis Complex and Their Association with Drug Resistance, 2nd ed.; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Pan, J.; Li, X.; Zhang, M.; Lu, Y.; Zhu, Y.; Wu, K.; Wu, Y.; Wang, W.; Chen, B.; Liu, Z.; et al. TransFlow: A Snakemake Workflow for Transmission Analysis of Mycobacterium tuberculosis Whole-Genome Sequencing Data. Bioinformatics 2023, 39, btac785. [Google Scholar] [CrossRef] [PubMed]
- Jandrasits, C.; Kröger, S.; Haas, W.; Renard, B.Y. Computational Pan-Genome Mapping and Pairwise SNP-Distance Improve Detection of Mycobacterium tuberculosis Transmission Clusters. PLoS Comput. Biol. 2019, 15, e1007527. [Google Scholar] [CrossRef]
- Cox, H.; Goig, G.A.; Salaam-Dreyer, Z.; Dippenaar, A.; Reuter, A.; Mohr-Holland, E.; Daniels, J.; Cudahy, P.G.T.; Nicol, M.P.; Borrell, S.; et al. Whole-Genome Sequencing Has the Potential to Improve Treatment for Rifampicin-Resistant Tuberculosis in High-Burden Settings: A Retrospective Cohort Study. J. Clin. Microbiol. 2022, 60, e0236221. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Ködmön, C. Whole-Genome Sequencing as Tool for Investigating International Tuberculosis Outbreaks: A Systematic Review. Front. Public Health 2019, 7, 87. [Google Scholar] [CrossRef]
- Hatherell, H.-A.; Colijn, C.; Stagg, H.R.; Jackson, C.; Winter, J.R.; Abubakar, I. Interpreting Whole Genome Sequencing for Investigating Tuberculosis Transmission: A Systematic Review. BMC Med. 2016, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Séraphin, M.N.; Norman, A.; Rasmussen, E.M.; Gerace, A.M.; Chiribau, C.B.; Rowlinson, M.-C.; Lillebaek, T.; Lauzardo, M. Direct Transmission of Within-Host Mycobacterium tuberculosis Diversity to Secondary Cases Can Lead to Variable between-Host Heterogeneity without de Novo Mutation: A Genomic Investigation. EBioMedicine 2019, 47, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; Lee, R.S.; Cowley, L.A.; Gardy, J.L.; Hanage, W.P. Within-Host Mycobacterium tuberculosis Diversity and Its Utility for Inferences of Transmission. Microb. Genom. 2018, 4, e000217. [Google Scholar] [CrossRef] [PubMed]
- Conceição, E.C.; dos Santos Guimarães, A.E.; Lopes, M.L.; Furlaneto, I.P.; Rodrigues, Y.C.; da Conceição, M.L.; Barros, W.A.; Cardoso, N.C.; Sharma, A.; Lima, L.N.G.C.; et al. Analysis of Potential Household Transmission Events of Tuberculosis in the City of Belem, Brazil. Tuberculosis 2018, 113, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Oostvogels, S.; Ley, S.D.; Heupink, T.H.; Dippenaar, A.; Streicher, E.M.; De Vos, E.; Meehan, C.J.; Dheda, K.; Warren, R.; Van Rie, A. Transmission, Distribution and Drug Resistance-Conferring Mutations of Extensively Drug-Resistant Tuberculosis in the Western Cape Province, South Africa. Microb. Genom. 2022, 8, 000815. [Google Scholar] [CrossRef]
- Cancino-Muñoz, I.; López, M.G.; Torres-Puente, M.; Villamayor, L.M.; Borrás, R.; Borrás-Máñez, M.; Bosque, M.; Camarena, J.J.; Colijn, C.; Colomer-Roig, E.; et al. Population-Based Sequencing of Mycobacterium tuberculosis Reveals How Current Population Dynamics Are Shaped by Past Epidemics. eLife 2022, 11, e76605. [Google Scholar] [CrossRef] [PubMed]
- Villalva-Serra, K.; Barreto-Duarte, B.; Miguez-Pinto, J.P.; Queiroz, A.T.L.; Rodrigues, M.M.; Rebeiro, P.F.; Amorim, G.; Cordeiro-Santos, M.; Sterling, T.R.; Araújo-Pereira, M.; et al. Impact of Xpert MTB/RIF Implementation in Tuberculosis Case Detection and Control in Brazil: A Nationwide Intervention Time-Series Analysis (2011–2022). Lancet Reg. Health–Am. 2024, 36. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Hasnain, S.E.; O’Toole, R.F.; Grover, S.; Ehtesham, N.Z. Whole Genome Sequencing: A New Paradigm in the Surveillance and Control of Human Tuberculosis. Tuberculosis 2015, 95, 91–94. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Sample ID | Isolation Year | Mapped Reads 1 (%) | Number Mapped Reads 1 | Median Coverage 1 | pDST 2 | gDST 3 | MIRU-VNTR Cluster | WGS Cluster (SNPs) 4 |
|---|---|---|---|---|---|---|---|---|
| G21377 | 2004 | 99.15 | 5,257,415 | 119 | MDR | MDR | Cluster 5 | <5 |
| G21379 | 2003 | 99.17 | 5,924,978 | 135 | MDR | MDR | Cluster 5 | <5 |
| G21383 | 2008 | 98.90 | 4,391,267 | 98 | MDR | MDR | Cluster 5 | <5 |
| G21389 | 2007 | 99.16 | 7,025,682 | 158 | MDR | MDR | Cluster 4 | <5 |
| G21376 | 2005 | 99.00 | 6,289,609 | 142 | MDR | MDR | Cluster 2 | >5 and <12 |
| G21382 | 2010 | 99.14 | 6,303,757 | 144 | MDR | MDR | Cluster 1 | >5 and <12 |
| G21384 | 2000 | 99.13 | 6,487,012 | 147 | MDR | MDR | Cluster 3 | >5 and <12 |
| G21385 | 2006 | 99.09 | 6,198,316 | 140 | MDR | MDR | Cluster 1 | >5 and <12 |
| G21386 | 2005 | 99.07 | 5,669,689 | 128 | MDR | MDR | Cluster 1 | >5 and <12 |
| G21388 | 2007 | 99.11 | 6,528,434 | 148 | MDR | MDR | Cluster 2 | >5 and <12 |
| G21391 | 2006 | 98.96 | 6,003,285 | 135 | MDR | MDR | Cluster 3 | >5 and <12 |
| G21392 | 2008 | 98.02 | 6,177,468 | 119 | MDR | MDR | Cluster 4 | >5 and <12 |
| G21393 | 2007 | 98.78 | 7,010,293 | 156 | MDR | MDR | Cluster 2 | >5 and <12 |
| G21387 | 2010 | 99.03 | 7,018,299 | 158 | MDR | MDR | No clustered | >12 |
| G21390 | 2005 | 98.96 | 7,763,638 | 171 | MDR | Pre-XDR | Cluster 2 | >12 |
| NP_1694 | 2003 | No data | No data | No data | MDR | No data | Cluster 3 | No data |
| NP_2392 | 2009 | No data | No data | No data | MDR | No data | Cluster 3 | No data |
| NP_2399 | 2007 | No data | No data | No data | MDR | No data | Cluster 2 | No data |
| NP_2522 | 2008 | No data | No data | No data | S | No data | Cluster 3 | No data |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conceição, E.C.; Loubser, J.; Guimarães, A.E.d.S.; Sharma, A.; Rutaihwa, L.K.; Dippenaar, A.; Salvato, R.S.; de Paula Souza e Guimarães, R.J.; da Silva Lourenço, M.C.; Barros, W.A.; et al. A Genome-Focused Investigation Reveals the Emergence of a Mycobacterium tuberculosis Strain Related to Multidrug-Resistant Tuberculosis in the Amazon Region of Brazil. Microorganisms 2024, 12, 1817. https://doi.org/10.3390/microorganisms12091817
Conceição EC, Loubser J, Guimarães AEdS, Sharma A, Rutaihwa LK, Dippenaar A, Salvato RS, de Paula Souza e Guimarães RJ, da Silva Lourenço MC, Barros WA, et al. A Genome-Focused Investigation Reveals the Emergence of a Mycobacterium tuberculosis Strain Related to Multidrug-Resistant Tuberculosis in the Amazon Region of Brazil. Microorganisms. 2024; 12(9):1817. https://doi.org/10.3390/microorganisms12091817
Chicago/Turabian StyleConceição, Emilyn Costa, Johannes Loubser, Arthur Emil dos Santos Guimarães, Abhinav Sharma, Liliana Kokusanilwa Rutaihwa, Anzaan Dippenaar, Richard Steiner Salvato, Ricardo José de Paula Souza e Guimarães, Maria Cristina da Silva Lourenço, Wandyra Araújo Barros, and et al. 2024. "A Genome-Focused Investigation Reveals the Emergence of a Mycobacterium tuberculosis Strain Related to Multidrug-Resistant Tuberculosis in the Amazon Region of Brazil" Microorganisms 12, no. 9: 1817. https://doi.org/10.3390/microorganisms12091817