
Gestational Interrelationships among Gut–Metabolism–Transcriptome in Regulating Early Embryo Implantation and Placental Development in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Consent
2.2. Animals and Samples Collection
2.3. RNA-Seq and Differential Gene Expression Analysis
2.4. Microbial DNA Extraction and 16S rRNA Gene Sequencing
2.5. Nontargeted Serum Metabolite Profiling and Analysis
2.6. Random Forest Model for Selection Pregnant Markers
2.7. Statistical Analysis
3. Results
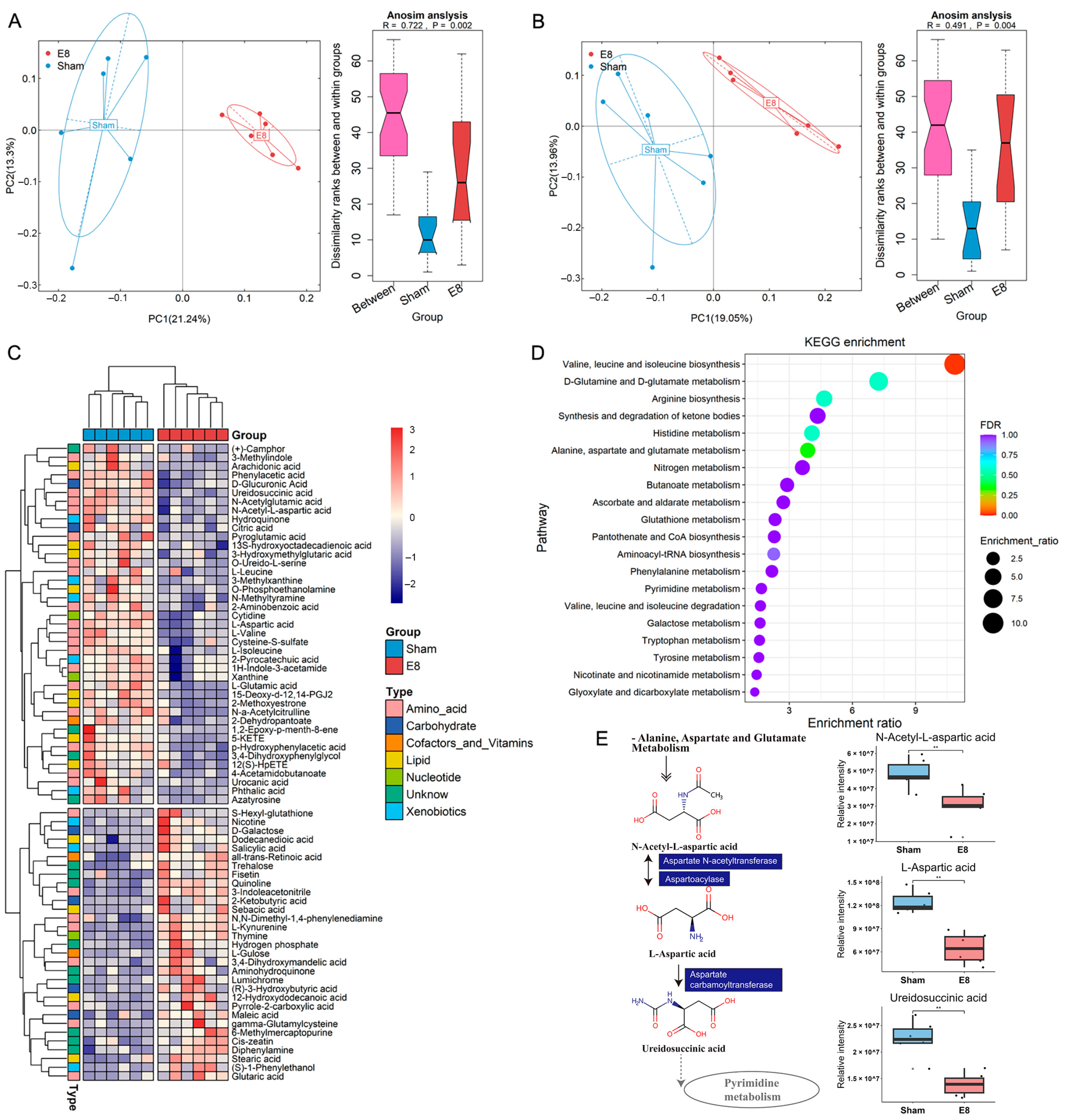
3.1. Early Pregnant Mice Show a Distinct Serum Metabolic Profile Compared with Mice in Sham Group
3.2. Gut Microbiota Composition Was Altered in E8 Mice
3.3. The Transcriptome of Gravid Uterus in E8 Mice Was Distinct Compared to Un-Pregnant Mice
3.4. The Putative Mechanistic Correlations among Serum Metabolites, Gut Microbiota and Uterus Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Hsueh, Y.W.; Chang, C.W.; Hsu, H.C.; Yang, T.C.; Lin, W.C.; Chang, H.M. Establishment of the fetal-maternal interface: Developmental events in human implantation and placentation. Front. Cell Dev. Biol. 2023, 11, 1200330. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Investig. 2004, 114, 744–754. [Google Scholar] [CrossRef]
- Akaeda, S.; Aikawa, S.; Hirota, Y. Spatial and molecular anatomy of the endometrium during embryo implantation: A current overview of key regulators of blastocyst invasion. FEBS J. 2024. [Google Scholar] [CrossRef]
- Boldeanu, L.; Dijmarescu, A.L.; Radu, M.; Silosi, C.A.; Popescu-Driga, M.V.; Poenariu, I.S.; Silosi, I.; Boldeanu, M.V.; Novac, M.B.; Novac, L.V. The role of mediating factors involved in angiogenesis during implantation. Rom. J. Morphol. Embryol. 2020, 61, 665–672. [Google Scholar] [CrossRef]
- Gurner, K.H.; Truong, T.T.; Harvey, A.J.; Gardner, D.K. A combination of growth factors and cytokines alter preimplantation mouse embryo development, foetal development and gene expression profiles. Mol. Hum. Reprod. 2020, 26, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Fuhler, G.M. The immune system and microbiome in pregnancy. Best Pract. Res. Clin. Gastroenterol. 2020, 44–45, 101671. [Google Scholar] [CrossRef] [PubMed]
- Nyangahu, D.D.; Jaspan, H.B. Influence of maternal microbiota during pregnancy on infant immunity. Clin. Exp. Immunol. 2019, 198, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; de Aguero, M.G.; Ganal-Vonarburg, S.C. How nutrition and the maternal microbiota shape the neonatal immune system. Nat. Rev. Immunol. 2017, 17, 508–517. [Google Scholar] [CrossRef]
- Giannella, L.; Grelloni, C.; Quintili, D.; Fiorelli, A.; Montironi, R.; Alia, S.; Delli Carpini, G.; Di Giuseppe, J.; Vignini, A.; Ciavattini, A. Microbiome Changes in Pregnancy Disorders. Antioxidants 2023, 12, 463. [Google Scholar] [CrossRef]
- Groer, M.W.; Gregory, K.E.; Louis-Jacques, A.; Thibeau, S.; Walker, W.A. The very low birth weight infant microbiome and childhood health. Birth Defects Res. Part C Embryo Today 2015, 105, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Siena, M.; Laterza, L.; Matteo, M.V.; Mignini, I.; Schepis, T.; Rizzatti, G.; Ianiro, G.; Rinninella, E.; Cintoni, M.; Gasbarrini, A. Gut and Reproductive Tract Microbiota Adaptation during Pregnancy: New Insights for Pregnancy-Related Complications and Therapy. Microorganisms 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Backhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial Changes during Pregnancy, Birth, and Infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.S.; Lu, J.H.; Li, S.H.; Li, J.H.; Yuan, M.Y.; He, J.R.; Chen, N.N.; Xiao, W.Q.; Shen, S.Y.; Qiu, L.; et al. Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Moylan, H.E.C.; Nguyen-Ngo, C.; Lim, R.; Lappas, M. The short-chain fatty acids butyrate and propionate protect against inflammation-induced activation of mediators involved in active labor: Implications for preterm birth. Mol. Hum. Reprod. 2020, 26, 452–468. [Google Scholar] [CrossRef]
- Antony, K.M.; Ma, J.; Mitchell, K.B.; Racusin, D.A.; Versalovic, J.; Aagaard, K. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am. J. Obstet. Gynecol. 2015, 212, 653.e1–653.e16. [Google Scholar] [CrossRef]
- Lu, X.; Shi, Z.; Jiang, L.; Zhang, S. Maternal gut microbiota in the health of mothers and offspring: From the perspective of immunology. Front. Immunol. 2024, 15, 1362784. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Oresic, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018, 149, 3–12. [Google Scholar] [CrossRef]
- Xie, G.; Zhang, S.; Zheng, X.; Jia, W. Metabolomics approaches for characterizing metabolic interactions between host and its commensal microbes. Electrophoresis 2013, 34, 2787–2798. [Google Scholar] [CrossRef]
- Handelman, S.K.; Romero, R.; Tarca, A.L.; Pacora, P.; Ingram, B.; Maymon, E.; Chaiworapongsa, T.; Hassan, S.S.; Erez, O.J.P.O. The plasma metabolome of women in early pregnancy differs from that of non-pregnant women. PLoS ONE 2019, 14, e0224682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Francis, E.; Hu, G.; Chen, L. Metabolomic profiling of women with gestational diabetes mellitus and their offspring: Review of metabolomics studies. J. Diabetes Complicat. 2018, 32, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Enquobahrie, D.A.; Denis, M.; Tadesse, M.G.; Gelaye, B.; Ressom, H.W.; Williams, M.A. Maternal Early Pregnancy Serum Metabolites and Risk of Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2015, 100, 4348–4356. [Google Scholar] [CrossRef] [PubMed]
- Kuc, S.; Koster, M.P.; Pennings, J.L.; Hankemeier, T.; Berger, R.; Harms, A.C.; Dane, A.D.; Schielen, P.C.; Visser, G.H.; Vreeken, R.J. Metabolomics profiling for identification of novel potential markers in early prediction of preeclampsia. PLoS ONE 2014, 9, e98540. [Google Scholar] [CrossRef]
- Jasarevic, E.; Bale, T.L. Prenatal and postnatal contributions of the maternal microbiome on offspring programming. Front. Neuroendocrinol. 2019, 55, 100797. [Google Scholar] [CrossRef]
- Vuong, H.E.; Pronovost, G.N.; Williams, D.W.; Coley, E.J.L.; Siegler, E.L.; Qiu, A.; Kazantsev, M.; Wilson, C.J.; Rendon, T.; Hsiao, E.Y. The maternal microbiome modulates fetal neurodevelopment in mice. Nature 2020, 586, 281–286. [Google Scholar] [CrossRef]
- Zhu, Y.; Mao, H.; Peng, G.; Zeng, Q.; Wei, Q.; Ruan, J.; Huang, J. Effect of JAK-STAT pathway in regulation of fatty liver hemorrhagic syndrome in chickens. Anim. Biosci. 2021, 34, 143–153. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Liu, Q.; He, M.; Zeng, Z.; Huang, X.; Fang, S.; Zhao, Y.; Ke, S.; Wu, J.; Zhou, Y.; Xiong, X.; et al. Extensive identification of serum metabolites related to microbes in different gut locations and evaluating their associations with porcine fatness. Microb. Biotechnol. 2023, 16, 1293–1311. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Di Simone, N.; Santamaria Ortiz, A.; Specchia, M.; Tersigni, C.; Villa, P.; Gasbarrini, A.; Scambia, G.; D’Ippolito, S. Recent Insights on the Maternal Microbiota: Impact on Pregnancy Outcomes. Front. Immunol. 2020, 11, 528202. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Pradhan, P.; Mallott, E.K.; Shirola, W.; Lu, A. Host-gut microbiota interactions during pregnancy. Evol. Med. Public Health 2024, 12, 7–23. [Google Scholar] [CrossRef]
- Pronovost, G.N.; Yu, K.B.; Coley-O’Rourke, E.J.L.; Telang, S.S.; Chen, A.S.; Vuong, H.E.; Williams, D.W.; Chandra, A.; Rendon, T.K.; Paramo, J.; et al. The maternal microbiome promotes placental development in mice. Sci. Adv. 2023, 9, eadk1887. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Lee-Sarwar, K.; Kelly, R.S.; Lasky-Su, J.A.; Litonjua, A.A.; Weiss, S.T.; Liu, Y.Y. Revealing the importance of prenatal gut microbiome in offspring neurodevelopment in humans. EBioMedicine 2023, 90, 104491. [Google Scholar] [CrossRef]
- Zhao, R.; An, Z.; Sun, Y.; Xia, L.; Qiu, L.; Yao, A.; Liu, Y.; Liu, L. Metabolic profiling in early pregnancy and associated factors of folate supplementation: A cross-sectional study. Clin. Nutr. 2021, 40, 5053–5061. [Google Scholar] [CrossRef]
- Barchitta, M.; Maugeri, A.; Magnano San Lio, R.; Favara, G.; La Mastra, C.; La Rosa, M.C.; Agodi, A. Dietary Folate Intake and Folic Acid Supplements among Pregnant Women from Southern Italy: Evidence from the “Mamma & Bambino” Cohort. Int. J. Environ. Res. Public Health 2020, 17, 638. [Google Scholar] [CrossRef]
- Yang, J.; Wu, J.; Tekola-Ayele, F.; Li, L.J.; Bremer, A.A.; Lu, R.; Rahman, M.L.; Weir, N.L.; Pang, W.W.; Chen, Z.; et al. Plasma Amino Acids in Early Pregnancy and Midpregnancy and Their Interplay with Phospholipid Fatty Acids in Association with the Risk of Gestational Diabetes Mellitus: Results from a Longitudinal Prospective Cohort. Diabetes Care 2023, 46, 722–732. [Google Scholar] [CrossRef]
- Stegink, L.D. Absorption, utilization, and safety of aspartic acid. J. Toxicol. Environ. Health 1976, 2, 215–242. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M. Aspartic Acid in Health and Disease. Nutrients 2023, 15, 4023. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Galvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef]
- Dong, L.; Du, H.; Zhang, M.; Xu, H.; Pu, X.; Chen, Q.; Luo, R.; Hu, Y.; Wang, Y.; Tu, H.; et al. Anti-inflammatory effect of Rhein on ulcerative colitis via inhibiting PI3K/Akt/mTOR signaling pathway and regulating gut microbiota. Phytother. Res. 2022, 36, 2081–2094. [Google Scholar] [CrossRef]
- Pereira, F.C.; Wasmund, K.; Cobankovic, I.; Jehmlich, N.; Herbold, C.W.; Lee, K.S.; Sziranyi, B.; Vesely, C.; Decker, T.; Stocker, R.; et al. Rational design of a microbial consortium of mucosal sugar utilizers reduces Clostridiodes difficile colonization. Nat. Commun. 2020, 11, 5104. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Sun, Y.; Xu, Y.; Jin, P.; Ding, H.; Dong, M. Case report: A compound heterozygous mutations in ASNS broadens the spectrum of asparagine synthetase deficiency in the prenatal diagnosis. Front. Pediatr. 2023, 11, 1273789. [Google Scholar] [CrossRef]
- Wang, C.; He, G.; Ge, Y.; Li, R.; Li, Z.; Lin, Y. A novel compound heterozygous missense mutation in ASNS broadens the spectrum of asparagine synthetase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1235. [Google Scholar] [CrossRef]
- Schleinitz, D.; Seidel, A.; Stassart, R.; Klammt, J.; Hirrlinger, P.G.; Winkler, U.; Kohler, S.; Heiker, J.T.; Schonauer, R.; Bialek, J.; et al. Novel Mutations in the Asparagine Synthetase Gene (ASNS) Associated with Microcephaly. Front. Genet. 2018, 9, 245. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Liang, Y.; Geng, J.; Yan, Y.; Ding, R.; He, M. Gestational Interrelationships among Gut–Metabolism–Transcriptome in Regulating Early Embryo Implantation and Placental Development in Mice. Microorganisms 2024, 12, 1902. https://doi.org/10.3390/microorganisms12091902
Lin S, Liang Y, Geng J, Yan Y, Ding R, He M. Gestational Interrelationships among Gut–Metabolism–Transcriptome in Regulating Early Embryo Implantation and Placental Development in Mice. Microorganisms. 2024; 12(9):1902. https://doi.org/10.3390/microorganisms12091902
Chicago/Turabian StyleLin, Shuai, Yuqi Liang, Jingqi Geng, Yunfei Yan, Ruipei Ding, and Maozhang He. 2024. "Gestational Interrelationships among Gut–Metabolism–Transcriptome in Regulating Early Embryo Implantation and Placental Development in Mice" Microorganisms 12, no. 9: 1902. https://doi.org/10.3390/microorganisms12091902
APA StyleLin, S., Liang, Y., Geng, J., Yan, Y., Ding, R., & He, M. (2024). Gestational Interrelationships among Gut–Metabolism–Transcriptome in Regulating Early Embryo Implantation and Placental Development in Mice. Microorganisms, 12(9), 1902. https://doi.org/10.3390/microorganisms12091902