
A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Bacteria
2.2. Identification of Bacterial Strains Through Real-Time PCR
2.3. Whole-Genome Sequencing and Annotations
2.4. Core Genome Multilocus Sequence Typing (cgMLST)
2.5. Clustering Analyses, Cluster Definition and Data Availability
2.6. Acquisition of Metadata
3. Results
3.1. Collection of Isolates and Molecular Identification
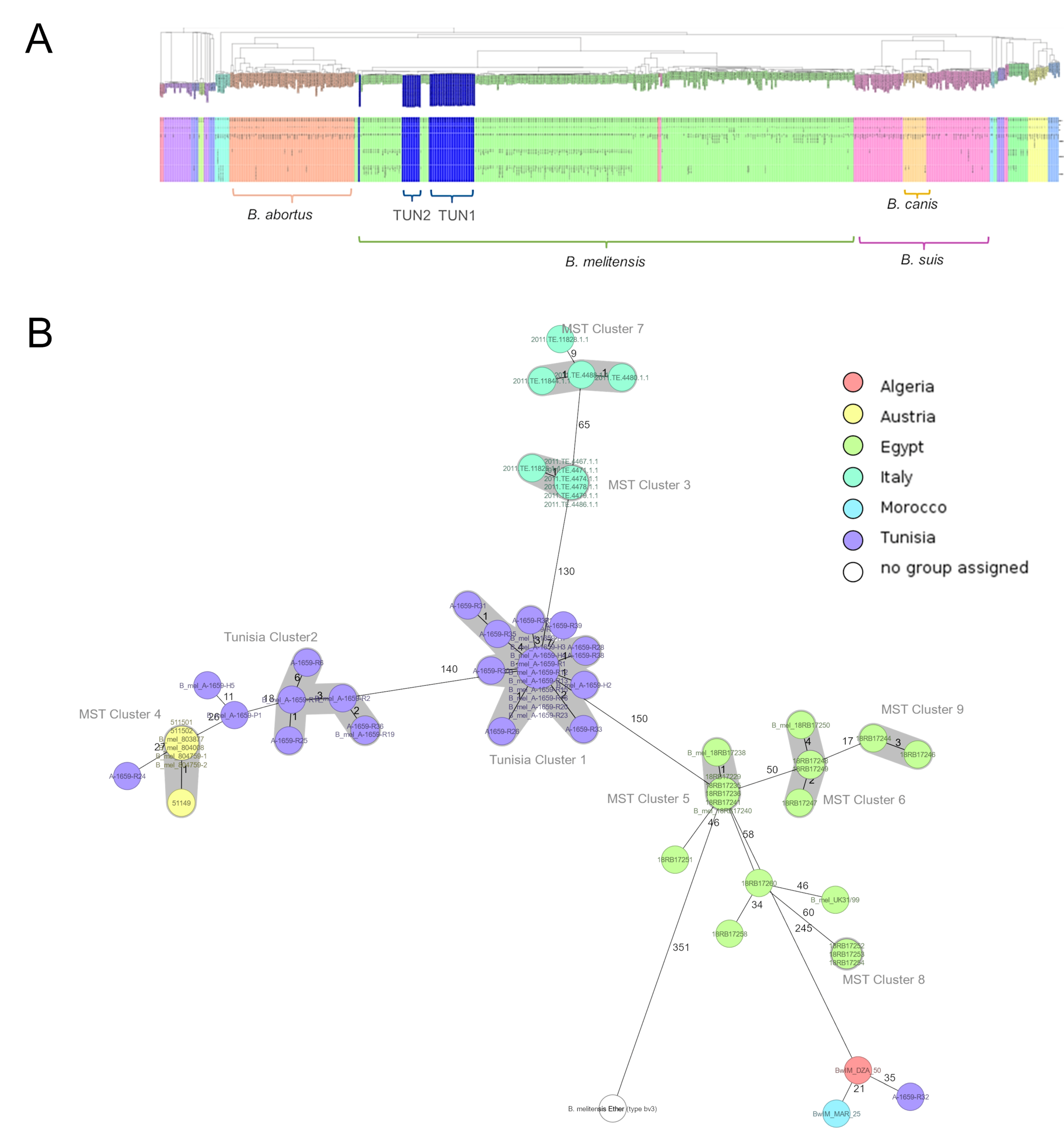
3.2. Whole-Genome Analysis and Core Genome Multilocus Sequence Typing (cgMLST)
3.3. Identification of AMR and Virulence Genes from WGS Data
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pappas, G.; Papadimitriou, P.; Akritidis, N.; Christou, L.; Tsianos, E.V. The new global map of human brucellosis. Lancet Infect Dis. 2006, 6, 91–99. [Google Scholar] [CrossRef]
- Lounes, N.; Cherfa, M.-A.; Le Carrou, G.; Bouyoucef, A.; Jay, M.; Garin-Bastuji, B.; Mick, V. Human Brucellosis in Maghreb: Existence of a Lineage Related to Socio-Historical Connections with Europe. PLoS ONE 2014, 9, e115319. [Google Scholar] [CrossRef]
- Franc, K.A.; Krecek, R.C.; Häsler, B.N.; Arenas-Gamboa, A.M. Brucellosis remains a neglected disease in the developing world: A call for interdisciplinary action. BMC Public Health 2018, 18, 125. [Google Scholar] [CrossRef]
- DSSB (Direction des Soins et de Santé de Base). Ministère de la Santé, Bulletin Epidémiologique. Annual Report; DSSB: Tunis, Tunisia, 2017.
- De Massis, F.; Di Girolamo, A.; Petrini, A.; Pizzigallo, E.; Giovannini, A. Correlation between animal and human brucellosis in Italy during the period 1997–2002. Clin. Microbiol. Infect. 2005, 11, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Gao, J.; Wu, Z. Laboratory-acquired infections with Brucella bacteria in China. Biosaf. Health 2021, 3, 101–104. [Google Scholar] [CrossRef]
- Hoover, D.L.; Friedlander, A. Brucellosis. In Textbook of Military Medicine: Medical Aspects of Chemical and Biological Warfare; Zajtchuk, R., Ed.; US Department of the Army, Surgeon General, and the Borden Institute: Washington, DC, USA, 1997. [Google Scholar]
- Olsen, S.C.; Boggiatto, P.; White, D.M.; McNunn, T. Biosafety Concerns Related to Brucella and Its Potential Use as a Bioweapon. Appl. Biosaf. J. ABSA Int. 2018, 23, 77–90. [Google Scholar] [CrossRef]
- Yagupsky, P.; Morata, P.; Colmenero, J.D. Laboratory Diagnosis of Human Brucellosis. Clin. Microbiol. Rev. 2020, 33, e00073-19. [Google Scholar] [CrossRef] [PubMed]
- Al Dahouk, S.; Scholz, H.C.; Tomaso, H.; Bahn, P.; Gollner, C.; Karges, W.; Appel, B.; Hensel, A.; Neubauer, H.; Nockler, K. Differential phenotyping of Brucella species using a newly developed semi-automated metabolic system. BMC Microbiol 2010, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Spink, W.W.; Hall, J.W.; Finstad, J.; Mallet, E. Immunization with viable Brucella organisms. Results of a safety test in humans. Bull. World Health Organ. 1962, 26, 409–419. [Google Scholar]
- Whatmore, A.M. Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 2009, 9, 1168–1184. [Google Scholar] [CrossRef]
- Corbel, M.J. Brucellosis: An overview. Emerg. Infect Dis. 1997, 3, 213–221. [Google Scholar] [CrossRef]
- Franco, M.P.; Mulder, M.; Gilman, R.H.; Smits, H.L. Human brucellosis. Lancet Infect Dis. 2007, 7, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Geresu, M.A.; Kassa, G.M. A Review on Diagnostic Methods of Brucellosis. J. Vet. Sci. Technol. 2016, 7, 3. [Google Scholar]
- Al Dahouk, S.; Tomaso, H.; Nöckler, K.; Neubauer, H.; Frangoulidis, D. Laboratory-Based Diagnosis of Brucellosis—A Review of the Literature. Part I: Techniques for Direct Detection and Identification of Brucella spp. Clin. Lab. 2003, 49, 487–505. [Google Scholar] [PubMed]
- Schaeffer, J.; Revilla-Fernández, S.; Hofer, E.; Posch, R.; Stoeger, A.; Leth, C.; Schmoll, F.; Djordjevic, V.; Lakicevic, B.; Matovic, K.; et al. Tracking the origin of Austrian human brucellosis cases using whole genome sequencing. Front. Med. 2021, 8, 635547. [Google Scholar] [CrossRef] [PubMed]
- Arapović, J.; Špičić, S.; Ostojić, M.; Duvnjak, S.; Arapović, M.; Nikolić, J.; Cvetnić, Ž. Epidemiological, clinical and molecular characterization of human brucellosis in Bosnia and Herzegovina—An ongoing brucellosis outbreak. Acta Med. Acad. 2018, 47, 50–60. [Google Scholar] [CrossRef]
- Georgi, E.; Walter, M.C.; Pfalzgraf, M.T.; Northoff, B.H.; Holdt, L.M.; Scholz, H.C.; Antwerpen, M.H. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS ONE 2017, 12, e0175425. [Google Scholar] [CrossRef]
- De Massis, F.; Ali, R.M.; Serrani, S.; Toro, M.; Sferrella, A.; D’aurelio, N.; Janowicz, A.; Zilli, K.; Romualdi, T.; Felicioni, E.; et al. Genetic Diversity of Brucella melitensis Isolated from Domestic Ruminants in Iraq. Microorganisms 2024, 12, 475. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Andrews, P.J.; Citerio, G. Eurotherm3235trial. Intensive Care Med. 2010, 36, 1990–1992. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genomeassemblyalgorithm and its applications to single-cellsequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef]
- Janowicz, A.; De Massis, F.; Ancora, M.; Cammà, C.; Patavino, C.; Battisti, A.; Prior, K.; Harmsen, D.; Scholz, H.; Zilli, K.; et al. Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of Brucella melitensis infections. J. Clin. Microbiol. 2018, 56, e00517-18. [Google Scholar] [CrossRef]
- Refai, M. Incidence and control of brucellosis in the Near East region. Vet. Microbiol. 2002, 90, 81–110. [Google Scholar] [CrossRef]
- Bouzouaia, N.; Chakroun, M.; Rachdi, J.; Rachdi, T. Epidemiological, clinical and therapeutic aspects of brucellosis in Tunisia. Apropos of epidemics in Gafsa. Tunis Med. 1995, 73, 443–448. [Google Scholar]
- Godfroid, J. Brucellosis in livestock and wildlife: Zoonotic diseases without pandemic potential in need of innovative one health approaches. Arch. Public Health 2017, 75, 34. [Google Scholar] [CrossRef]
- Battikh, H.; Berriche, A.; Zayoud, R.; Ammari, L.; Abdelmalek, R.; Kilani, B.; Tiouiri Ben Aissa, H.; Zribi, M. Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure. Infect. Dis. Now. 2021, 51, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Cao, X.A.; Wang, M.; Piao, D.R.; Zhao, H.Y.; Cui, B.Y.; Jiang, H.; Li, Z.J. Whole-genome sequencing of a Brucella melitensis strain (BMWS93) isolated from a bank clerk and exhibiting complete resistance to rifampin. Microbiol. Resour. Announc. 2019, 8, e01645-01618. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene | Primers | Sequence (5′–3′) | Localization |
|---|---|---|---|
| mazG | mazG-F | GGATCTGATCGTAGCGACGGA | 3651–3671 |
| mazG-R | CGTCCAATGTCTCACTGGAAAA | 3751–3772 | |
| Bru-mazG-TM-Multi | FAM-TGCCTTACATGGGCGAACTCGAACGT-BHQ-1 | 3677–3703 | |
| IS711 | BruIS-F | GCCATCAGATTGAATGCTTTTTTAAC | 701–726 |
| BruIS-R | AACCAGATCATAGCGCATGCG | 801–821 | |
| Bru-IS-TM-Multi | Cy5-CGCTGCGATGCGAGAAAACATTGACC-BHQ-2 | 752–777 | |
| KoMa2 | KoMa-For | GGTGATGCCGCATTATTACTAGG | 198–220 |
| KoMa-Rev | GGTATTAGCAGTCGCAGGCTT | 336–316 | |
| KoMa-TM | HEX-TTCTTGCTTGAGGATCTGTCGTGGATCG-BHQ-2 | 224–251 |
| Temperatures [°C] | Time [s] | Cycles | Steps |
|---|---|---|---|
| 95 | 600 | Denaturing | |
| 95 | 15 | ×40 | Annealing Elongation + Detection |
| 60 | 60 |
| Isolate | Accesion | Year | Hospital | Origin | Age (Years) | Gender | Clinical Form | cgMLST |
|---|---|---|---|---|---|---|---|---|
| A-1659-R23 | SAMN38851271 | 2016 | Rabta | Béja | 37 | M | Bacteremia | |
| A-1659-H1 | SAMN38851272 | 2017 | CNH | Ben Arous | 45 | F | Bacteremia | |
| A-1659-H3 | SAMN38851273 | 2017 | CNH | Tunis | 42 | M | Bacteremia | |
| A-1659-H4 | SAMN38851274 | 2017 | CNH | Siliana | 37 | F | Bacteremia | |
| A-1659-R1 | SAMN38851275 | 2017 | Rabta | Tunis | 47 | M | Bacteremia | |
| A-1659-R3 | SAMN38851276 | 2017 | Rabta | Tunis | 55 | M | Bacteremia | |
| A-1659-R4 | SAMN38851277 | 2017 | Rabta | Zaghouen | 37 | F | Bacteremia | |
| A-1659-R12 | SAMN38851278 | 2017 | Rabta | Tunis | 41 | M | Bacteremia | |
| A-1659-R13 | SAMN38851279 | 2017 | Rabta | Nabeul | 28 | F | Bacteremia | |
| A-1659-R15 | SAMN38851280 | 2017 | Rabta | Tunis | 39 | M | Bacteremia | |
| A-1659-R16 | SAMN38851281 | 2017 | Rabta | Béja | 59 | M | Bacteremia | |
| A-1659-R18 | SAMN38851282 | 2017 | Rabta | Tunis | 37 | M | Bacteremia | |
| A-1659-R20 | SAMN38851283 | 2017 | Rabta | Tunis | 51 | M | Bacteremia | Tunisia |
| A-1659-R27 | SAMN38851284 | 2017 | Rabta | Béja | 22 | M | Bacteremia | Cluster 1 |
| A-1659-R31 | SAMN38851285 | 2017 | Rabta | Ben Arous * | 27 | M | Bacteremia | |
| A-1659-R33 | SAMN38851286 | 2017 | Rabta | Mannouba | 25 | M | Bacteremia ** | |
| A-1659-R35 | SAMN38851287 | 2017 | Rabta | Ben Arous | 21 | M | Bacteremia | |
| A-1659-R37 | SAMN38851288 | 2017 | Rabta | Tunis | 20 | F | Bacteremia | |
| A-1659-R38 | SAMN38851289 | 2017 | Rabta | Bizete | 30 | M | Bacteremia | |
| A-1659-R39 | SAMN38851290 | 2017 | Rabta | Tunis | 68 | M | Bacteremia | distant |
| A-1659-H2 | SAMN38851291 | 2017 | CNH | Béja | 35 | F | Bacteremia | |
| A-1659-R14 | SAMN38851292 | 2017 | Rabta | Bizerte | 48 | M | Bacteremia | |
| A-1659-R26 | SAMN38851293 | 2017 | Rabta | Tunis | 63 | M | Bacteremia | |
| A-1659-R28 | SAMN38851294 | 2017 | Rabta | Tunis | 33 | M | Bacteremia | |
| A-1659-R30 | SAMN38851295 | 2017 | Rabta | Tunis | 63 | M | Bacteremia | |
| A-1659-R36 | SAMN38851296 | 2017 | Rabta | Tunis | 47 | M | Bacteremia | |
| A-1659-H5 | SAMN38851297 | 2017 | CNH | Tunis | 23 | M | Bacteremia | distant |
| A-1659-R2 | SAMN38851298 | 2017 | Rabta | Seliana | 59 | M | Spondilodiscitis | |
| A-1659-R6 | SAMN38851299 | 2017 | Rabta | Béja | 71 | M | Bacteremia | |
| A-1659-R11 | SAMN38851300 | 2017 | Rabta | Tunis | 21 | M | Bacteremia | Tunisia |
| A-1659-R17 | SAMN38851301 | 2016 | Rabta | Béja | 22 | M | Bacteremia | Cluster 2 |
| A-1659-R19 | SAMN38851302 | 2017 | Rabta | Tunis | 25 | M | Bacteremia | |
| A-1659-R25 | SAMN38851303 | 2016 | Rabta | Ben Arous | 16 | M | Bacteremia | |
| A-1659-P1 | SAMN38851304 | 2020 | CNH | Tunis | 37 | F | Bacteremia | distant |
| A-1659-R24 | SAMN38851305 | 2016 | Rabta | Béja | 68 | M | Bacteremia | distant |
| A-1659-R32 | SAMN38851306 | 2016 | Rabta | Bizerte | 26 | M | Bacteremia | distant |
| Type | Virulence | AMR-Related |
|---|---|---|
| Gene | acpXL, bigA, bigB, bmaB/omaA, bmaC, BPE005, BPE043, BPE123, BPE275, bspA, bspB, bspC, bspE, bspF, bspL, btpA, btpB, bvrR, bvrS, cgs, fabZ, gmd, htrB, kdsA, kdsB, lpsA, lpsB/lpcC, lpxA, lpxB, lpxC, lpxD, lpxE, lpxK, manAoAg, manBcore, manCcore, manCoAg, per, pgm, pmm, ricA, sepA, vceA, vceC, virB1, virB10, virB11, virB12, virB2, virB3, virB4, virB5, virB6, virB7, virB8, virB9, waaA/kdtA, wbdA, wbkA, wbkB, wbkC, wboA, wbpL, wbpZ, wzm, wzt | bepC, bepD, bepE, bepF, bepG, mprF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferjani, A.; Buijze, H.; Kopprio, G.; Köhler, S.; Rehaiem, A.; Battikh, H.; Ammari, L.; Ferjani, S.; Kanzari, L.; Zribi, M.; et al. A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure. Microorganisms 2025, 13, 243. https://doi.org/10.3390/microorganisms13020243
Ferjani A, Buijze H, Kopprio G, Köhler S, Rehaiem A, Battikh H, Ammari L, Ferjani S, Kanzari L, Zribi M, et al. A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure. Microorganisms. 2025; 13(2):243. https://doi.org/10.3390/microorganisms13020243
Chicago/Turabian StyleFerjani, Asma, Hellen Buijze, Germán Kopprio, Susanne Köhler, Amel Rehaiem, Hajer Battikh, Lamia Ammari, Sana Ferjani, Lamia Kanzari, Meriam Zribi, and et al. 2025. "A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure" Microorganisms 13, no. 2: 243. https://doi.org/10.3390/microorganisms13020243
APA StyleFerjani, A., Buijze, H., Kopprio, G., Köhler, S., Rehaiem, A., Battikh, H., Ammari, L., Ferjani, S., Kanzari, L., Zribi, M., Kilani, B., Hanschmann, N., Scholz, H., & Boutiba, I. (2025). A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure. Microorganisms, 13(2), 243. https://doi.org/10.3390/microorganisms13020243