
Microbiota Involved in the Degradation of Tremella fuciformis Polysaccharide and Microbial Enzymatic Potential Revealed by Microbiome and Metagenome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extraction of Tremella fuciformis Polysaccharides
2.3. Bacterial Screening and Purification
2.4. 16S rDNA Amplicon Sequencing
2.5. Metagenome Sequencing and Functional Annotation
2.6. Determination of Macro-Transcriptomics and Differential Expression Analysis
3. Results and Discussion
3.1. Enrichment of Microbiota from Different Sources
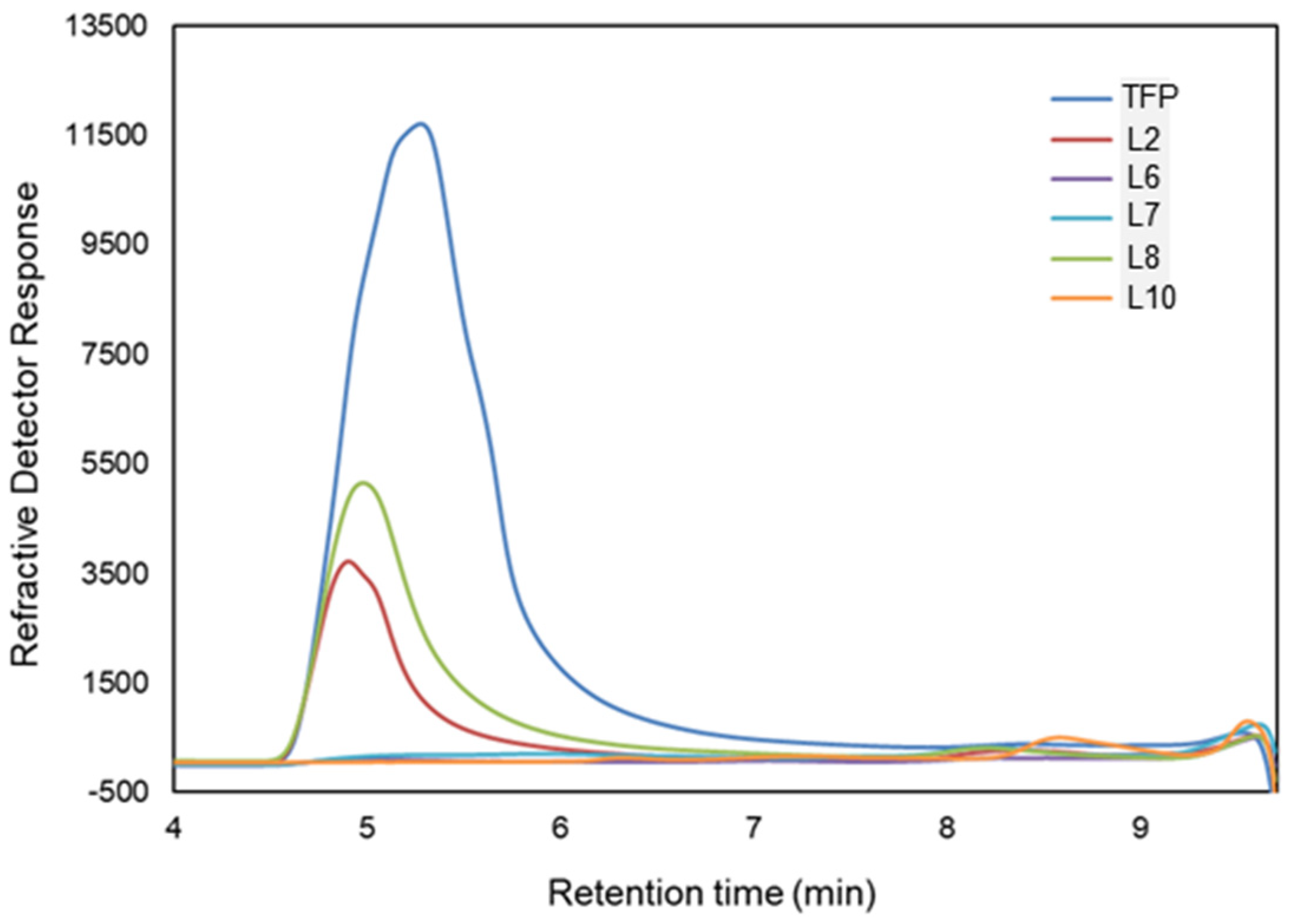
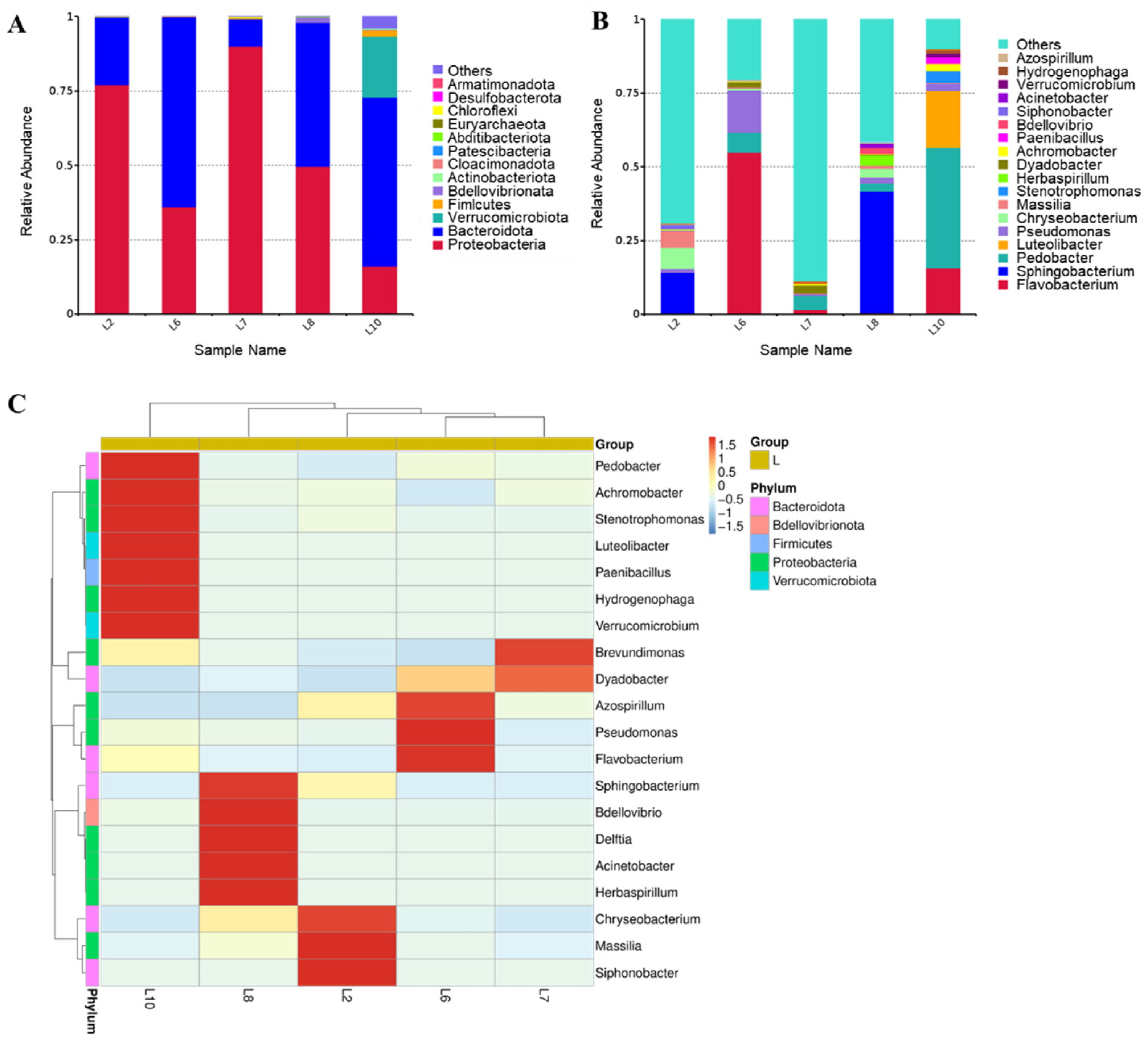
3.2. Enzymatic Activity and Bacterial Structure Analysis of Bacteria Capable of Utilizing TFP
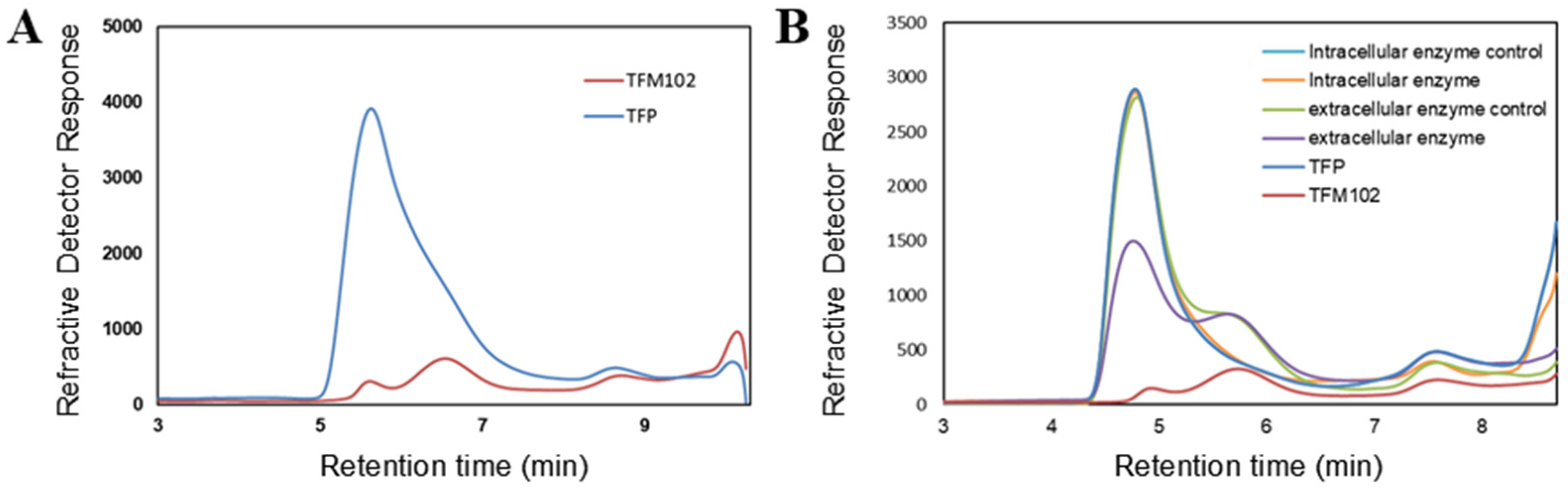
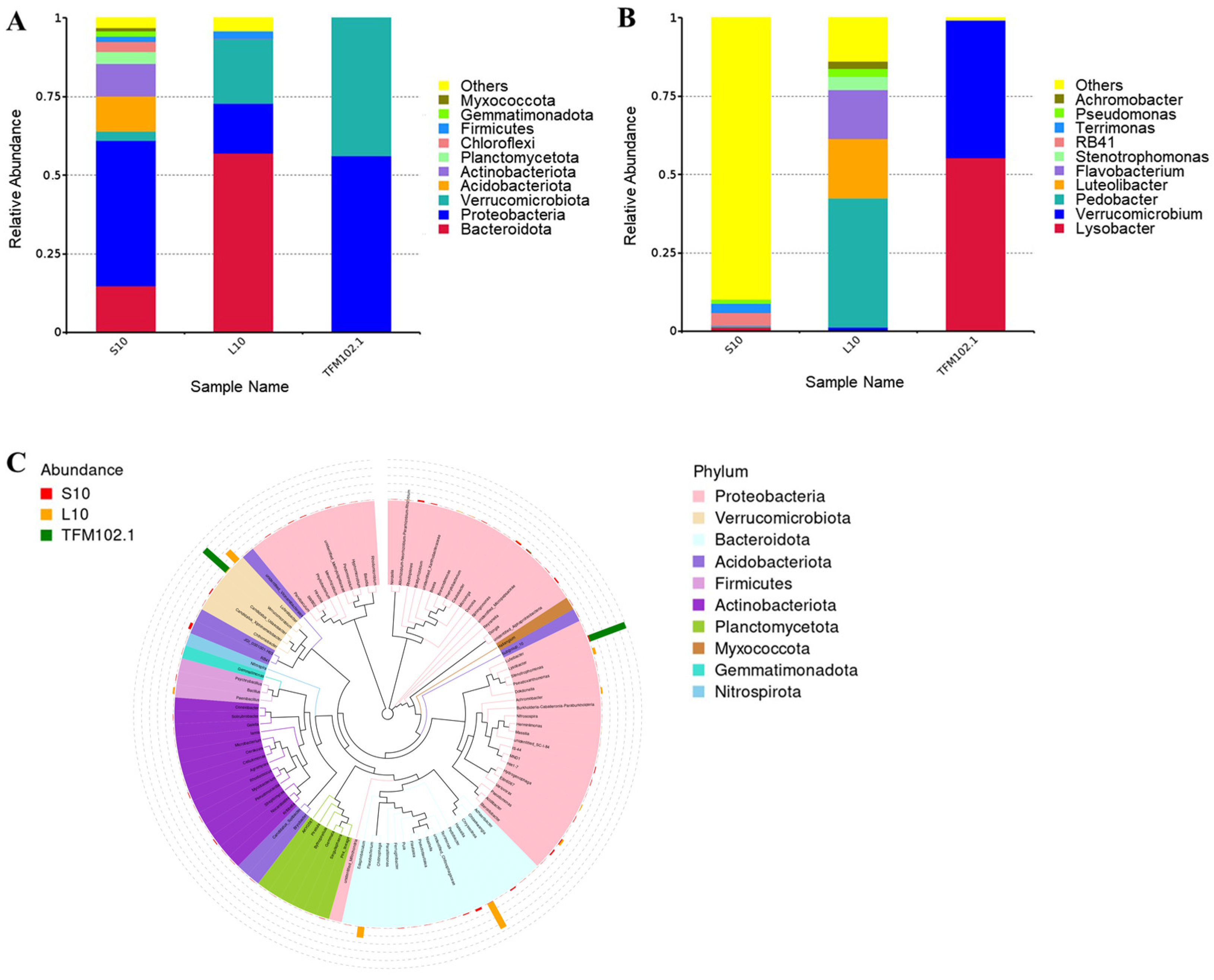
3.3. Metagenome Function Prediction of the Degrading Bacteriophage TFM102
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ebersberger, I.; de Matos Simoes, R.; Kupczok, A.; Gube, M.; Kothe, E.; Voigt, K.; von Haeseler, A. A Consistent Phylogenetic Backbone for the Fungi. Mol. Biol. Evol. 2011, 29, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, A.; Ge, X.; Li, S.; Zhang, T.; Xu, H. Chain conformation and physicochemical properties of polysaccharide (glucuronoxylomannan) from Fruit Bodies of Tremella fuciformis. Carbohydr. Polym. 2020, 245, 116354. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, X.-Z.; Bai, F.-Y. Four new species of Tremella (Tremellales, Basidiomycota) based on morphology and DNA sequence data. MycoKeys 2019, 47, 75–95. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, L.; Zhang, Z.; Zhang, W.; Tang, J.; He, X.; Zhou, J.; Peng, W. Tremella fuciformis Polysaccharides Inhibited Colonic Inflammation in Dextran Sulfate Sodium-Treated Mice via Foxp3+ T Cells, Gut Microbiota, and Bacterial Metabolites. Front. Immunol. 2021, 12, 648162. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Chen, T.; Huang, L.; Zhang, Y.; Feng, Y.; Qu, S.; Yin, X.; Liang, L.; Yan, J.; Liu, W. Tremella fuciformis polysaccharide reduces obesity in high-fat diet-fed mice by modulation of gut microbiota. Front. Microbiol. 2022, 13, 1073350. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Gao, Q.; Ma, C.-W.; Ge, Y.; You, L.; Liu, R.H.; Fu, X.; Liu, D. Effect of polysaccharides from Tremella fuciformis on UV-induced photoaging. J. Funct. Foods 2016, 20, 400–410. [Google Scholar] [CrossRef]
- Shen, T.; Duan, C.; Chen, B.; Li, M.; Ruan, Y.; Xu, D.; Shi, D.; Yu, D.; Li, J.; Wang, C. Tremella fuciformis polysaccharide suppresses hydrogen peroxide-triggered injury of human skin fibroblasts via upregulation of SIRT1. Mol. Med. Rep. 2017, 16, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-T.; An, L.-Y.; Liu, W.; Hu, Y.-C.; Wang, S.-P.; Zou, L. In vitro fecal fermentation properties of polysaccharides from Tremella fuciformis and related modulation effects on gut microbiota. Food Res. Int. 2022, 156, 111185. [Google Scholar] [CrossRef]
- Lee, Q.; Xue, Z.; Luo, Y.; Lin, Y.; Lai, M.; Xu, H.; Liu, B.; Zheng, M.; Lv, F.; Zeng, F. Low molecular weight polysaccharide of Tremella fuciformis exhibits stronger antioxidant and immunomodulatory activities than high molecular weight polysaccharide. Int. J. Biol. Macromol. 2024, 281, 136097. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, D.; Xu, J.; Tu, D.; Zhuang, W.; Tian, Y. Effect of polysaccharide concentration on heat-induced Tremella fuciformis polysaccharide-soy protein isolation gels: Gel properties and interactions. Int. J. Biol. Macromol. 2024, 262, 129782. [Google Scholar] [CrossRef]
- Tu, J.; Adhikari, B.; Brennan, M.A.; Cheng, P.; Bai, W.; Brennan, C.S. Interactions between sorghum starch and mushroom polysaccharides and their effects on starch gelatinization and digestion. Food Hydrocoll. 2023, 139, 108504. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, X.; Ma, M.; Long, T.; Xiao, C.; Zhang, J.; Liu, J.; Zhao, L. Immunoenhancing glucuronoxylomannan from Tremella aurantialba Bandoni et Zang and its low-molecular-weight fractions by radical depolymerization: Properties, structures and effects on macrophages. Carbohydr. Polym. 2020, 238, 116184. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Hu, X.; Zhang, Y.A.N.; Liu, T. Studies on the purification of polysaccharides separated from Tremella fuciformis and their neuroprotective effect. Mol. Med. Rep. 2016, 13, 3985–3992. [Google Scholar] [CrossRef] [PubMed]
- Rosselló-Mora, R.; Amann, R. The species concept for prokaryotes. FEMS Microbiol Rev. 2001, 25, 39–67. [Google Scholar] [CrossRef]
- Grondin Julie, M.; Tamura, K.; Déjean, G.; Abbott, D.W.; Brumer, H. Polysaccharide Utilization Loci: Fueling Microbial Communities. J. Bacteriol. 2017, 199, e00860-16. [Google Scholar] [CrossRef] [PubMed]
- Nacke, H.; Engelhaupt, M.; Brady, S.; Fischer, C.; Tautzt, J.; Daniel, R. Identification and characterization of novel cellulolytic and hemicellulolytic genes and enzymes derived from German grassland soil metagenomes. Biotechnol. Lett. 2011, 34, 663–675. [Google Scholar] [CrossRef]
- Verastegui, Y.; Cheng, J.; Engel, K.; Kolczynski, D.; Mortimer, S.; Lavigne, J.; Montalibet, J.; Romantsov, T.; Hall, M.; McConkey, B.J.; et al. Multisubstrate Isotope Labeling and Metagenomic Analysis of Active Soil Bacterial Communities. mBio 2014, 5, e01157-14. [Google Scholar] [CrossRef] [PubMed]
- Tláskal, V.; Baldrian, P. Deadwood-Inhabiting Bacteria Show Adaptations to Changing Carbon and Nitrogen Availability During Decomposition. Front. Microbiol. 2021, 12, 685303. [Google Scholar] [CrossRef]
- Costa, O.Y.A.; de Hollander, M.; Pijl, A.; Liu, B.; Kuramae, E.E. Cultivation-independent and cultivation-dependent metagenomes reveal genetic and enzymatic potential of microbial community involved in the degradation of a complex microbial polymer. Microbiome 2020, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Sime, A.M.; Kifle, B.A.; Woldesemayat, A.A.; Gemeda, M.T. Microbial carbohydrate active enzyme (CAZyme) genes and diversity from Menagesha Suba natural forest soils of Ethiopia as revealed by shotgun metagenomic sequencing. BMC Microbiol. 2024, 24, 285. [Google Scholar] [CrossRef]
- Pang, H.; Zhang, P.; Duan, C.-J.; Mo, X.-C.; Tang, J.-L.; Feng, J.-X. Identification of Cellulase Genes from the Metagenomes of Compost Soils and Functional Characterization of One Novel Endoglucanase. Curr. Microbiol. 2009, 58, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Chantorn, S.; Aekkawatchai, N.; Chunya, P.; Oontawee, S.; Khumphai, P.; Charoenrat, T. Lignocellulolytic bacteria isolated from organic rice field soils for enzyme production using agricultural wastes: Screening, medium optimization, and co-culture. Biocatal. Agric. Biotechnol. 2021, 33, 101988. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H.; Hancock, J. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Kuramae, E.E.; Conradie, T.A.; Jacobs, K. Distribution patterns of Acidobacteriota in different fynbos soils. PLoS ONE 2021, 16, e0248913. [Google Scholar]
- Nam, S.; Alday, J.G.; Kim, M.; Kim, H.; Kim, Y.; Park, T.; Lim, H.S.; Lee, B.Y.; Lee, Y.K.; Jung, J.Y. The relationships of present vegetation, bacteria, and soil properties with soil organic matter characteristics in moist acidic tundra in Alaska. Sci. Total Environ. 2021, 772, 145386. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.J.; Hahn, D.; Hengstmann, U.; Liesack, W.; Janssen, P.H. Characterization and identification of numerically abundant culturable bacteria from the anoxic bulk soil of rice paddy microcosms. Appl. Environ. Microbiol. 1999, 65, 5042–5049. [Google Scholar] [CrossRef] [PubMed]
- Domman, D.B.; Steven, B.T.; Ward, N.L. Random transposon mutagenesis of Verrucomicrobium spinosum DSM 4136T. Arch. Microbiol. 2010, 193, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Pold, G.; Conlon, E.M.; Huntemann, M.; Pillay, M.; Mikhailova, N.; Stamatis, D.; Reddy, T.B.K.; Daum, C.; Shapiro, N.; Kyrpides, N.; et al. Genome Sequence of Verrucomicrobium sp. Strain GAS474, a Novel Bacterium Isolated from Soil. Genome Announc. 2018, 6, e01451-17. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Jiang, J.; Taylor, A.J.; Leite, A.D.L.; Dodds, E.D.; Du, L. Outer Membrane Vesicle-Mediated Codelivery of the Antifungal HSAF Metabolites and Lytic Polysaccharide Monooxygenase in the Predatory Lysobacter enzymogenes. ACS Chem. Biol. 2021, 16, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Popper, Z.A.; Michel, G.; Hervé, C.; Domozych, D.S.; Willats, W.G.T.; Tuohy, M.G.; Kloareg, B.; Stengel, D.B. Evolution and Diversity of Plant Cell Walls: From Algae to Flowering Plants. Annu. Rev. Plant Biol. 2011, 62, 567–590. [Google Scholar] [CrossRef]
- Abot, A.; Arnal, G.; Auer, L.; Lazuka, A.; Labourdette, D.; Lamarre, S.; Trouilh, L.; Laville, E.; Lombard, V.; Potocki-Veronese, G.; et al. CAZyChip: Dynamic assessment of exploration of glycoside hydrolases in microbial ecosystems. Bmc Genom. 2016, 17, 671. [Google Scholar] [CrossRef]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Ge, X.Y.; Huang, W.W.; Xu, X.Q.; Lei, P.; Sun, D.F.; Xu, H.; Li, S. Production, structure, and bioactivity of polysaccharide isolated from Tremella fuciformis XY. Int. J. Biol. Macromol. 2020, 148, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, J.; Chang, Y.; Mei, X.; Luan, J.; Jiang, X.; Xue, C. The Discovery of a Multidomain Mannanase Containing Dual-Catalytic Domain of the Same Activity: Biochemical Properties and Synergistic Effect. J. Agric. Food Chem. 2024, 72, 10451–10458. [Google Scholar] [CrossRef]
- Liu, G.C.; Song, L.; Li, J.J.; Song, X.; Mei, X.W.; Zhang, Y.Y.; Fan, C.; Chang, Y.G.; Xue, C.H. Identification and characterization of a chondroitinase ABC with a novel carbohydrate-binding module. Int. J. Biol. Macromol. 2024, 271, 132518. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Chen, G.; Zheng, L.; Shen, J.; Xue, C.; Chang, Y. Microbiota Involved in the Degradation of Tremella fuciformis Polysaccharide and Microbial Enzymatic Potential Revealed by Microbiome and Metagenome. Microorganisms 2025, 13, 263. https://doi.org/10.3390/microorganisms13020263
Song X, Chen G, Zheng L, Shen J, Xue C, Chang Y. Microbiota Involved in the Degradation of Tremella fuciformis Polysaccharide and Microbial Enzymatic Potential Revealed by Microbiome and Metagenome. Microorganisms. 2025; 13(2):263. https://doi.org/10.3390/microorganisms13020263
Chicago/Turabian StyleSong, Xiao, Guangning Chen, Long Zheng, Jingjing Shen, Changhu Xue, and Yaoguang Chang. 2025. "Microbiota Involved in the Degradation of Tremella fuciformis Polysaccharide and Microbial Enzymatic Potential Revealed by Microbiome and Metagenome" Microorganisms 13, no. 2: 263. https://doi.org/10.3390/microorganisms13020263
APA StyleSong, X., Chen, G., Zheng, L., Shen, J., Xue, C., & Chang, Y. (2025). Microbiota Involved in the Degradation of Tremella fuciformis Polysaccharide and Microbial Enzymatic Potential Revealed by Microbiome and Metagenome. Microorganisms, 13(2), 263. https://doi.org/10.3390/microorganisms13020263