

Interpreting Microbial Species–Area Relationships: Effects of Sequence Data Processing Algorithms and Fitting Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Data Processing and Species Classification
2.3. The Combination of Species–Area Relationship Data
2.4. Linear Transformation and Fitting of the Power Model
2.5. Diversity Analysis and Visualization
2.6. Fitting and Selection of SAR Models
2.7. Visualization of the Best Models
3. Results
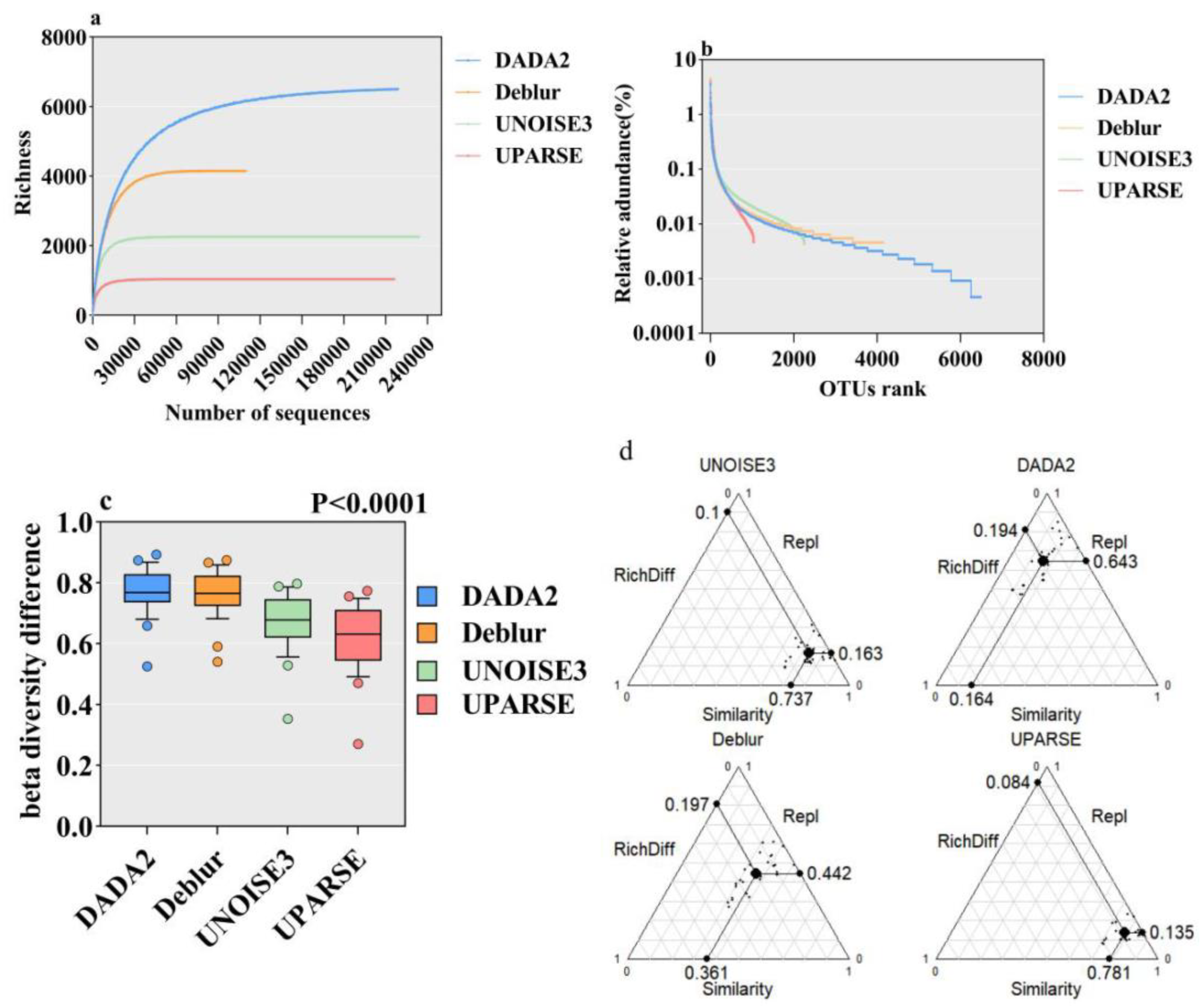
3.1. Impact of Different Algorithms on Microbial SARs and Species Diversity Comparison
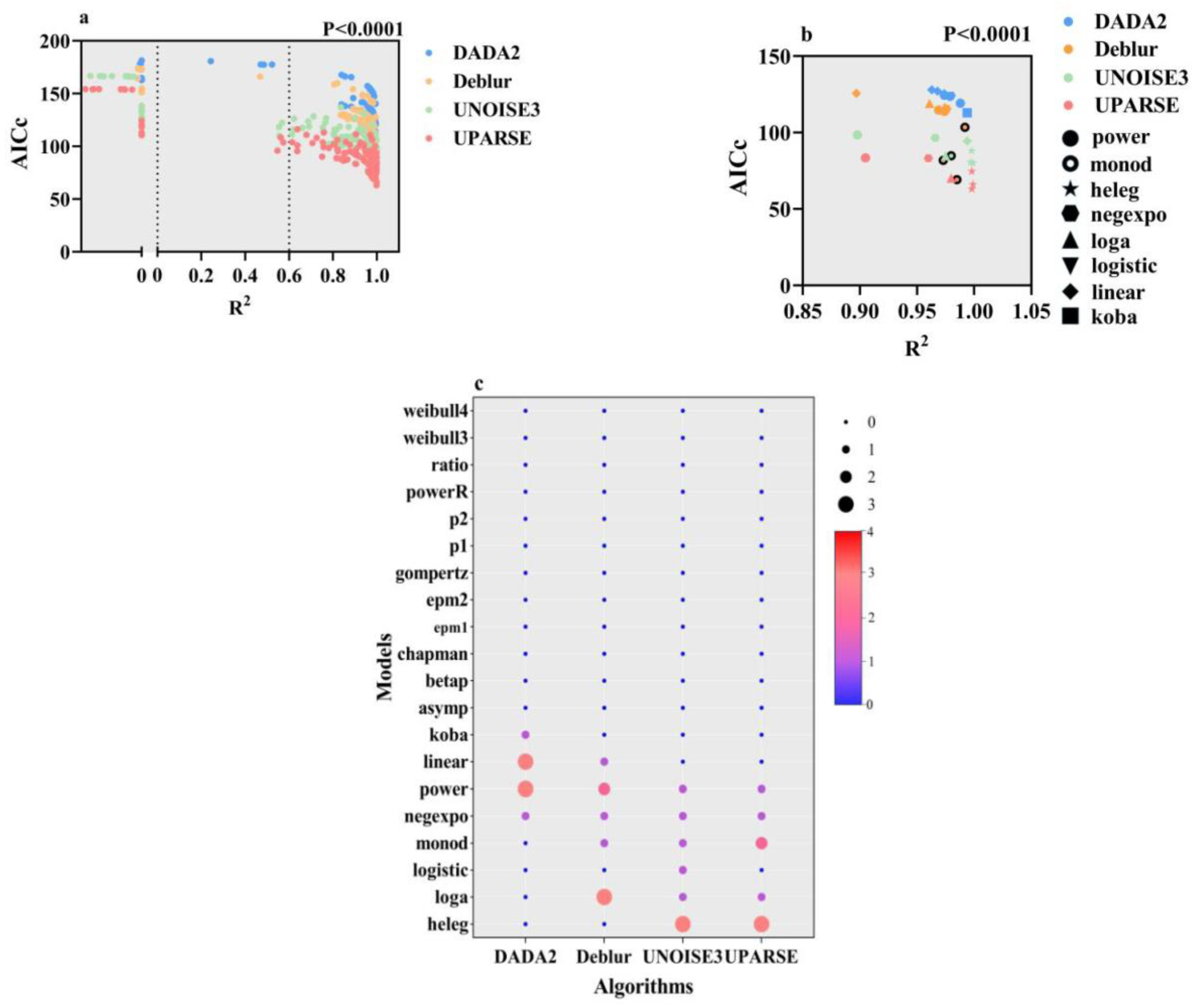
3.2. The Impact of Model Selection on SARs and the Compatibility Between Algorithms and Models
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Macarthur, R.H.; Wilson, E.O. An Equilibrium Theory of Insular Zoogeography. Evol. Int. J. Org. Evol. 1963, 17, 373–387. [Google Scholar] [CrossRef]
- Lomolino, M.V. Ecology’s most general, yet protean pattern: The species-area relationship. J. Biogeogr. 2000, 27, 17–26. [Google Scholar] [CrossRef]
- Tjørve, E.; Tjørve, K.M. Species–Area Relationship. In Encyclopedia of Life Sciences; Wiley: New York, NY, USA, 2017; pp. 1–9. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Bohannan, B.J.M. Spatial scaling of microbial biodiversity. Trends Ecol. Evol. 2006, 21, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yu, G.-B.; Yang, X.-Y.; Xiao, W. Testing the passive sampling hypothesis: The role of dispersal in shaping microbial species-area relationship. Front. Microbiol. 2023, 14, 1093695. [Google Scholar] [CrossRef]
- Li, S.P.; Wang, P.; Chen, Y.; Wilson, M.C.; Yang, X.; Ma, C.; Lu, J.; Chen, X.Y.; Wu, J.; Shu, W.S.; et al. Island biogeography of soil bacteria and fungi: Similar patterns, but different mechanisms. ISME J. 2020, 14, 1886–1896. [Google Scholar] [CrossRef]
- Moradi, H.; Fattorini, S.; Oldeland, J. Influence of elevation on the species–area relationship. J. Biogeogr. 2020, 47, 2029–2041. [Google Scholar] [CrossRef]
- Zhang, B.; Xue, K.; Liu, W.; Zhou, S.; Nie, S.; Rui, Y.; Tang, L.; Pang, Z.; Li, L.; Dong, J.; et al. Power law in species–area relationship overestimates bacterial diversity in grassland soils at larger scales. Glob. Ecol. Biogeogr. 2024, 33, e13825. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. MSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, M.; McCauley, M.; Villéger, S.; Jackson, C.R. Ranking the biases: The choice of OTUs vs. ASVs in 16S rRNA amplicon data analysis has stronger effects on diversity measures than rarefaction and OTU identity threshold. PLoS ONE 2022, 17, e0264443. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An independent evaluation of microbiome sequence error- correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef]
- Pan, X. Application of fundamental equations to species-area theory. BMC Ecol. 2016, 16, 42. [Google Scholar] [CrossRef]
- Drakare, S.; Lennon, J.J.; Hillebrand, H. The imprint of the geographical, evolutionary and ecological context on species-area relationships. Ecol. Lett. 2006, 9, 215–227. [Google Scholar] [CrossRef]
- Deng, W.; Cheng, Y.T.; Li, Z.Q.; Zhou, F.P.; Yang, X.Y.; Xiao, W. Passive sampling hypothesis did not shape microbial species-area relationships in open microcosm systems. Ecol. Evol. 2022, 12, e9634. [Google Scholar] [CrossRef]
- Deng, W.; Liu, L.L.; Yu, G.B.; Li, N.; Yang, X.Y.; Xiao, W. Testing the Resource Hypothesis of Species-Area Relationships: Extinction Cannot Work Alone. Microorganisms 2022, 10, 1993. [Google Scholar] [CrossRef] [PubMed]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Rosenzweig, M.L. Species Diversity in Space and Time; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Matthews, T.J.; Triantis, K.A.; Whittaker, R.J.; Guilhaumon, F. sars: An R package for fitting, evaluating and comparing species–area relationship models. Ecography 2019, 42, 1446–1455. [Google Scholar] [CrossRef]
- Møller, A.P.; Jennions, M.D. How much variance can be explained by ecologists and evolutionary biologists? Oecologia 2002, 132, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Burnham, K.P.; Anderson, D.R. Model Selection and Multi-Model Inferencs: A Practical Information-Theoretic Approach; Burnham, K.P., Anderson, D.R., Eds.; Springer: New York, NY, USA, 2010. [Google Scholar]
- Hurvich, C.M. Regression and time series model selection in small samples. Biometrika 1989, 76, 297–307. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Z.; Zhou, J.; Guo, X.; Chen, Y. Microbial Species-Area Relationships on the Skins of Amphibian Hosts. Microbiol. Spectr. 2023, 11, e0177122. [Google Scholar] [CrossRef]
- Ramos Sarmiento, K.; Carr, A.; Diener, C.; Locey, K.J.; Gibbons, S.M. Island biogeography theory provides a plausible explanation for why larger vertebrates and taller humans have more diverse gut microbiomes. ISME J. 2024, 18, wrae114. [Google Scholar] [CrossRef]
- Rivett, D.W.; Mombrikotb, S.B.; Gweon, H.S.; Bell, T.; van der Gast, C. Bacterial communities in larger islands have reduced temporal turnover. ISME J. 2021, 15, 2947–2955. [Google Scholar] [CrossRef]
- Xiao, W.; Ma, Z.S. Inter-Individual Diversity Scaling Analysis of the Human Virome With Classic Diversity-Area Relationship (DAR) Modeling. Front. Genet. 2021, 12, 627128. [Google Scholar] [CrossRef]
- Wang, P.; Li, S.P.; Yang, X.; Si, X.; Li, W.J.; Shu, W.; Jiang, L. Spatial scaling of soil microbial co-occurrence networks in a fragmented landscape. mLife 2023, 2, 209–215. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, F.-L.; Deng, W.; Cheng, Y.-T.; Yang, X.-Y.; Li, N.; Xiao, W. Interpreting Microbial Species–Area Relationships: Effects of Sequence Data Processing Algorithms and Fitting Models. Microorganisms 2025, 13, 635. https://doi.org/10.3390/microorganisms13030635
Qi F-L, Deng W, Cheng Y-T, Yang X-Y, Li N, Xiao W. Interpreting Microbial Species–Area Relationships: Effects of Sequence Data Processing Algorithms and Fitting Models. Microorganisms. 2025; 13(3):635. https://doi.org/10.3390/microorganisms13030635
Chicago/Turabian StyleQi, Fu-Liang, Wei Deng, Yi-Ting Cheng, Xiao-Yan Yang, Na Li, and Wen Xiao. 2025. "Interpreting Microbial Species–Area Relationships: Effects of Sequence Data Processing Algorithms and Fitting Models" Microorganisms 13, no. 3: 635. https://doi.org/10.3390/microorganisms13030635
APA StyleQi, F.-L., Deng, W., Cheng, Y.-T., Yang, X.-Y., Li, N., & Xiao, W. (2025). Interpreting Microbial Species–Area Relationships: Effects of Sequence Data Processing Algorithms and Fitting Models. Microorganisms, 13(3), 635. https://doi.org/10.3390/microorganisms13030635