
Pangenome Analysis of Clostridium scindens: A Collection of Diverse Bile Acid- and Steroid-Metabolizing Commensal Gut Bacterial Strains

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Sequences
2.2. Genome Annotation
2.3. Metagenome-Assembled Genomes (MAGs) of C. scindens from Public Metagenomes
2.4. Determination of the Pangenome of 34 C. scindens Strains
2.5. Distance Analysis of Strains
2.6. Functional Annotation: Prediction of COG and KEGG Groups
2.7. Identification of bai and des Genes in C. scindens Genomes
2.8. Phylogenomic Analysis
3. Results
3.1. Assembly and Genomic Characteristics of C. scindens Strains
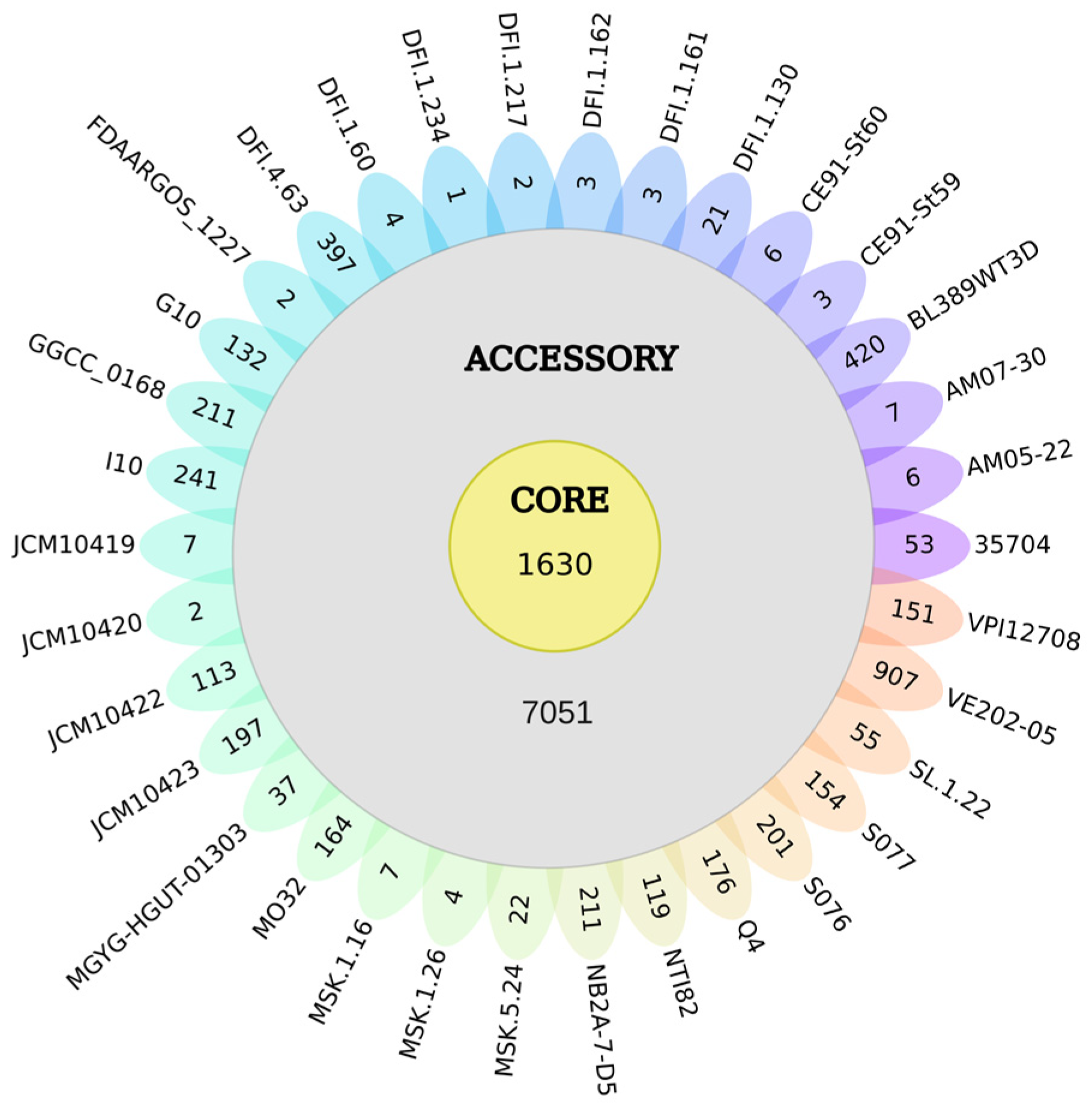
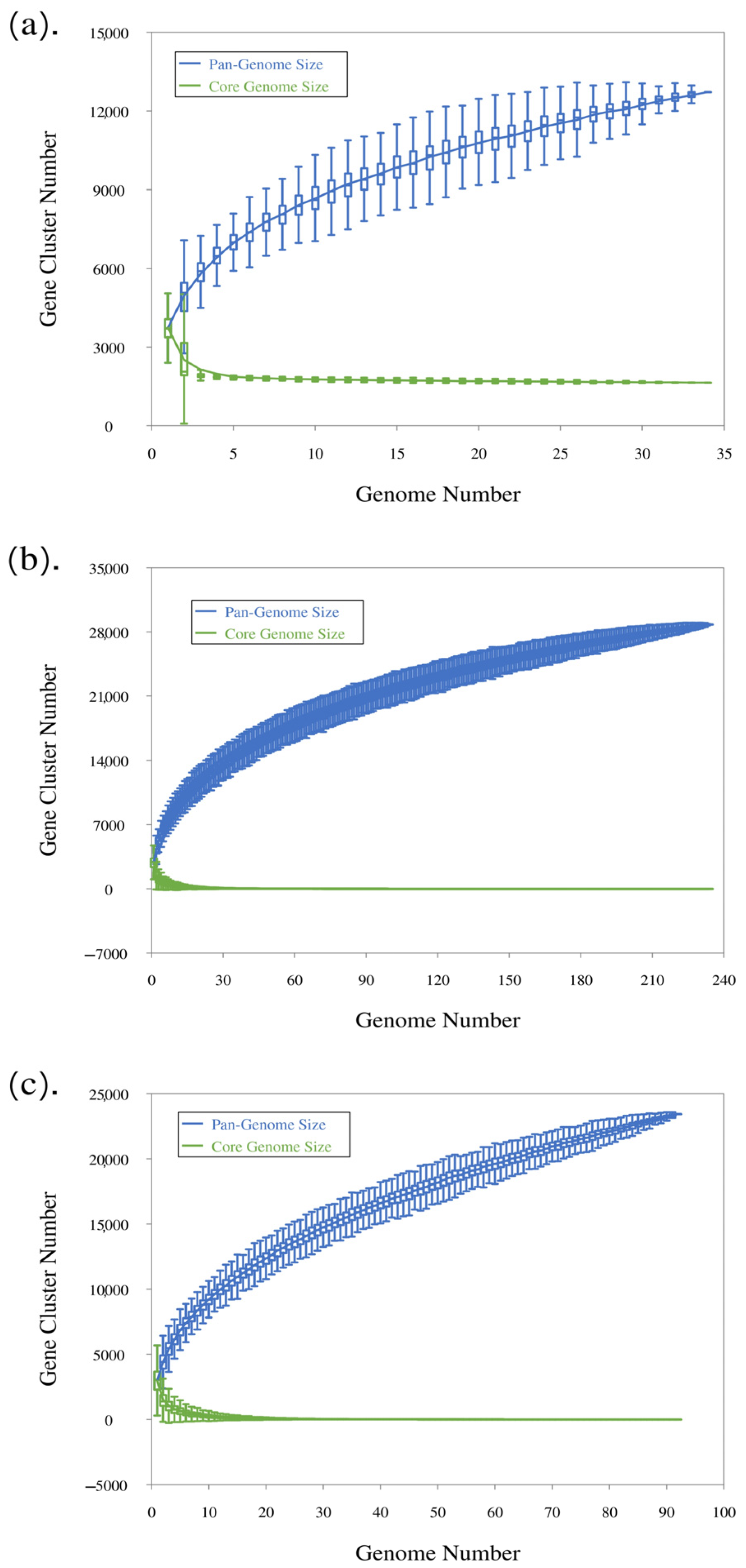
3.2. Pangenome of the 34 Cultured Strains of C. scindens
3.3. Pangenome Analysis After Addition of C. scindens Metagenome-Assembled Genomes
3.4. Pangenome Profile
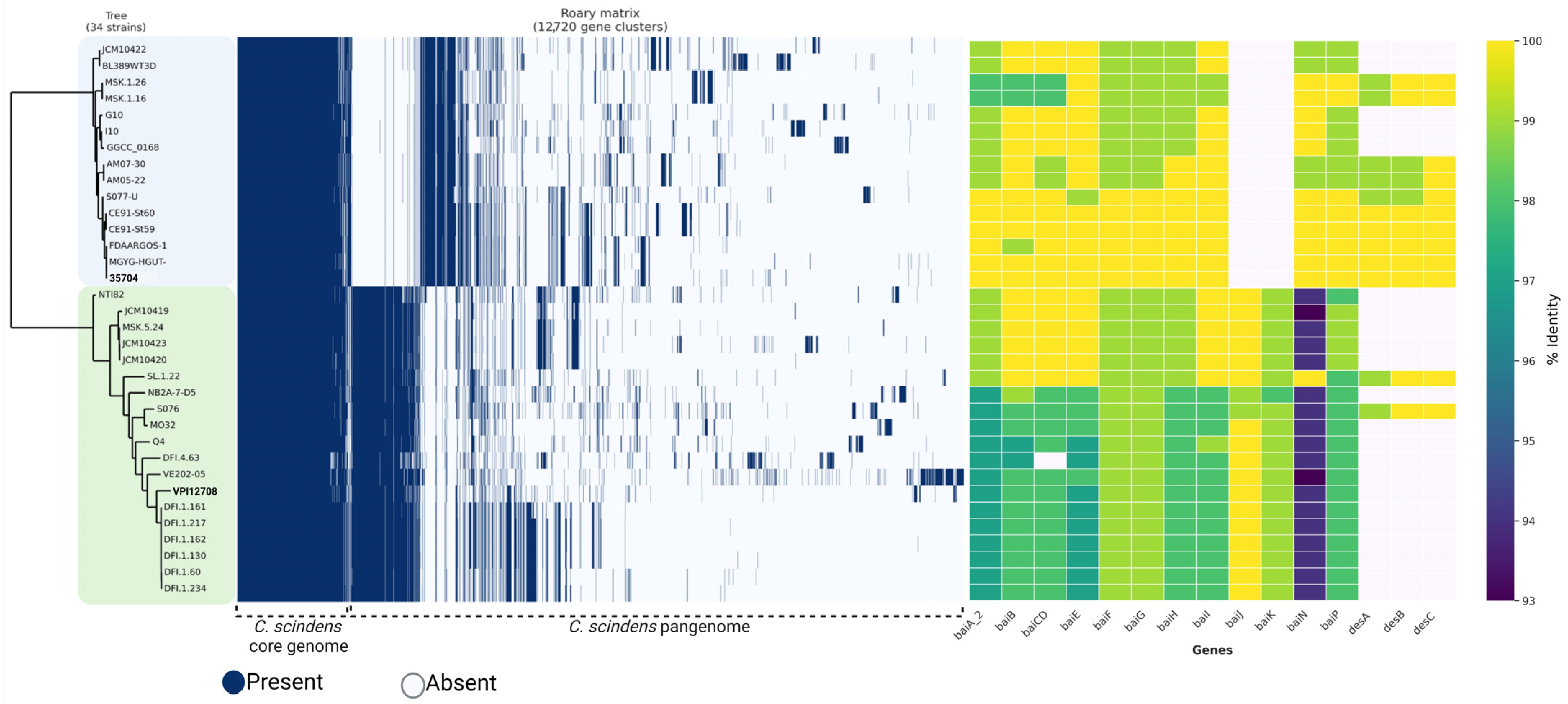
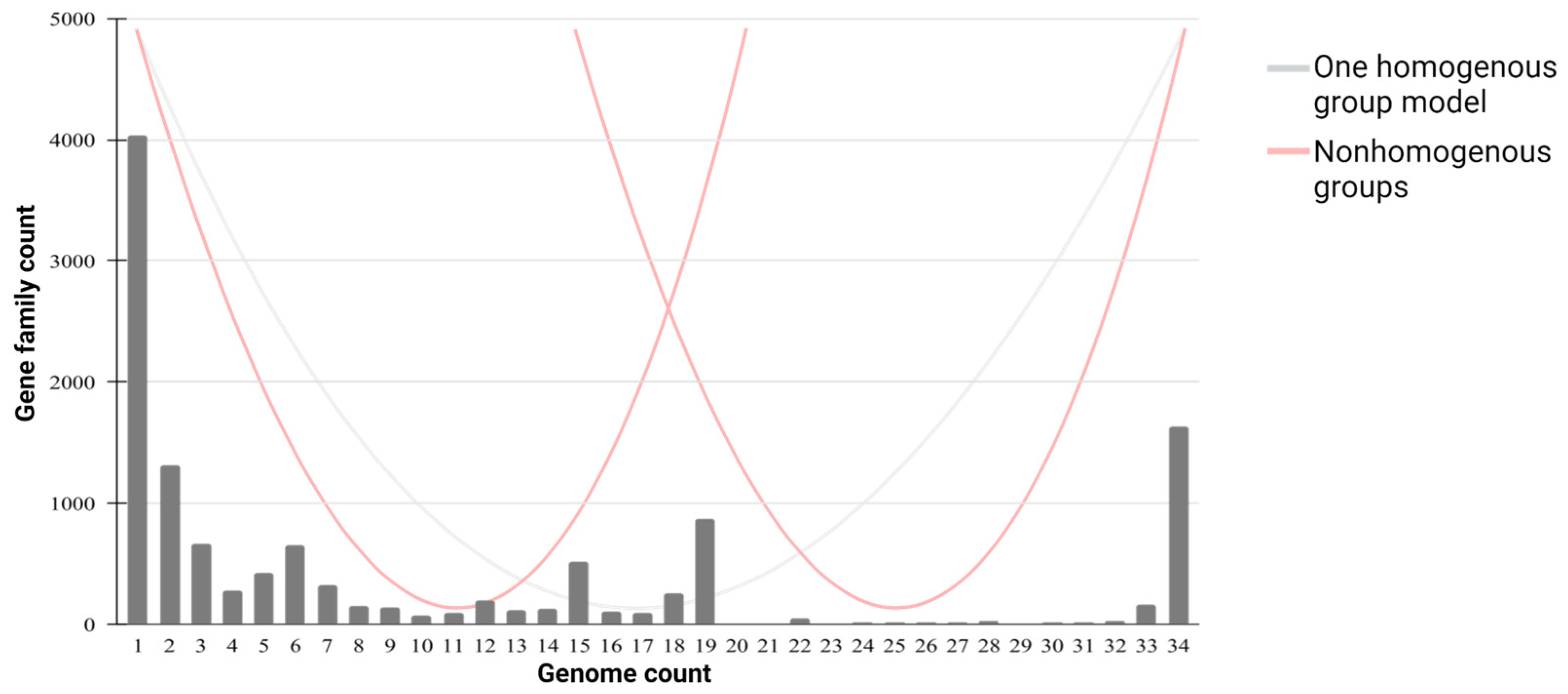
3.5. Identification of C. scindens Strain Groups
3.6. Average Nucleotide Identity Analysis of 34 C. scindens Strain Cultivars
3.7. SSU rDNA Analysis of 34 C. scindens Strain Cultivars
3.8. COG Distributions of C. scindens Core, Accessory, and Unique Genes
3.9. KEGG Pathway Distributions of the C. scindens Pangenome
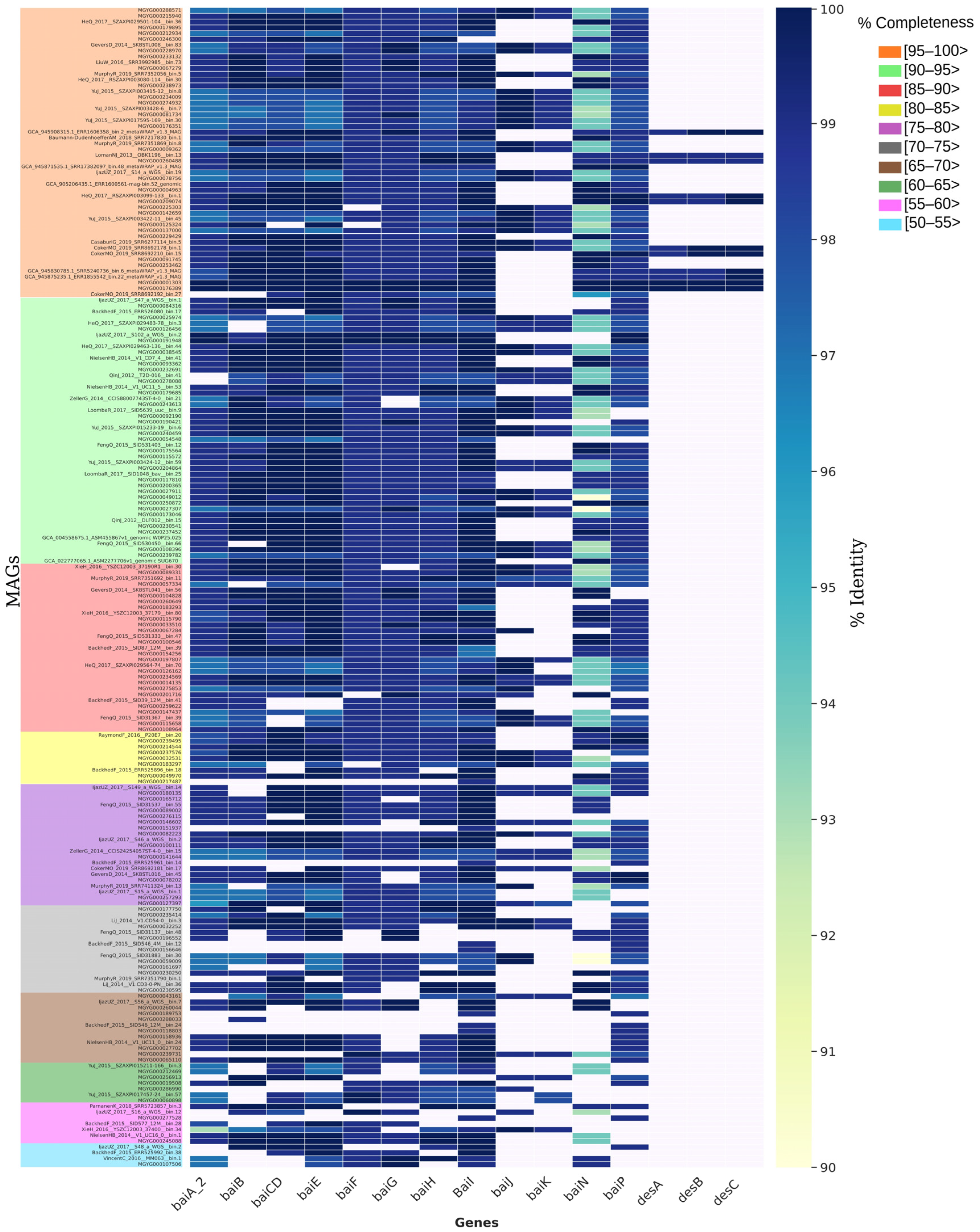
3.10. Identification of Bile-Acid-Metabolizing Genes in C. scindens Genomes
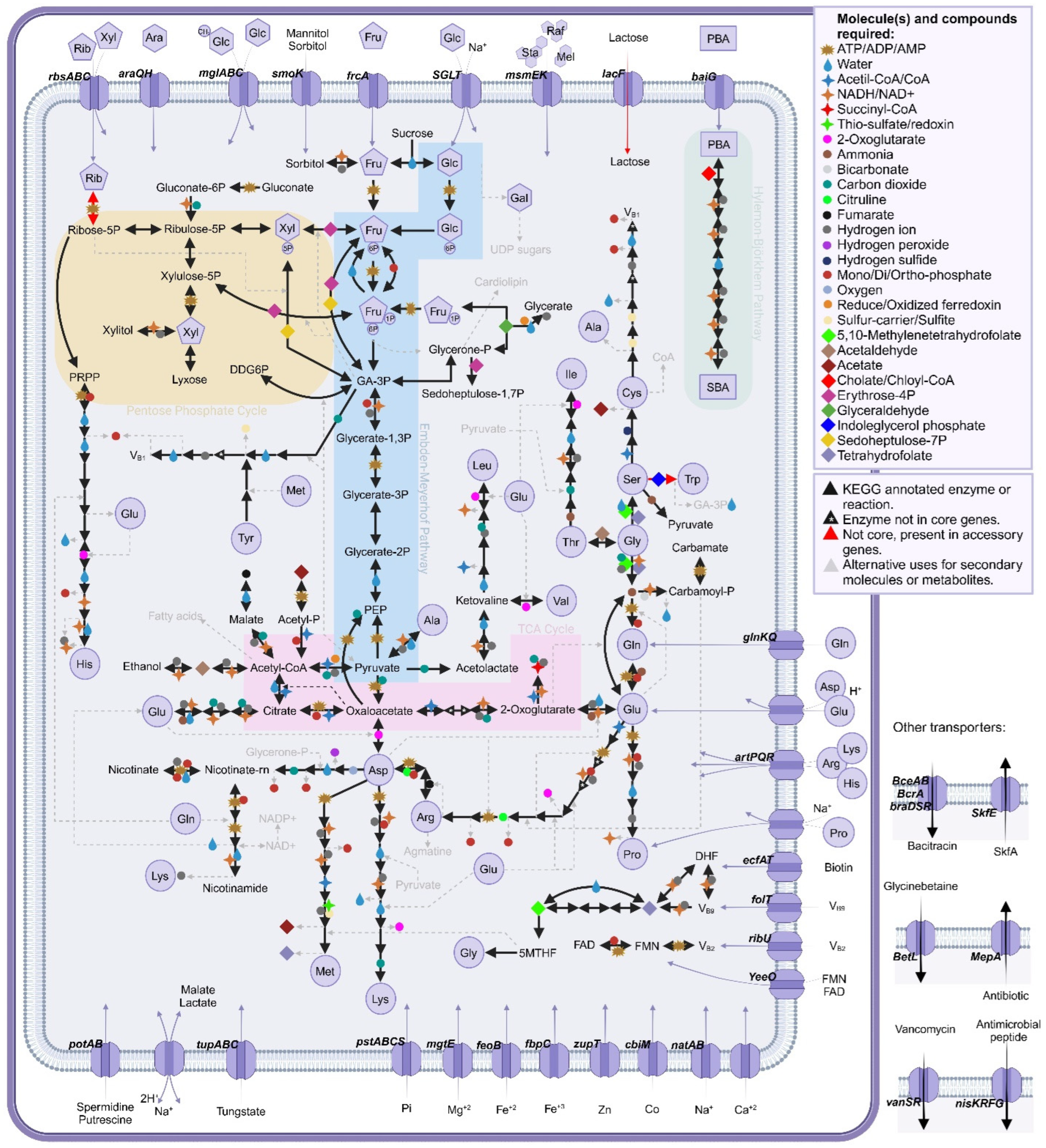
3.11. Predicted Metabolic Pathways in the Core Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Daniel, S.L.; Gaskins, H.R. The Hylemon-Bjorkhem pathway of bile acid 7-dehydroxylation: History, biochemistry, and microbiology. J. Lipid Res. 2023, 64, 100392. [Google Scholar] [CrossRef]
- Winter, J.; Morris, G.N.; O’Rourke-Locascio, S.; Bokkenheuser, V.D.; Mosbach, E.H.; Cohen, B.I.; Hylemon, P.B. Mode of action of steroid desmolase and reductases synthesized by Clostridium “scindens” (formerly Clostridium strain 19). J. Lipid Res. 1984, 25, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- White, B.A.; Lipsky, R.L.; Fricke, R.J.; Hylemon, P.B. Bile acid induction specificity of 7α-dehydroxylase activity in an intestinal Eubacterium species. Steroids 1980, 35, 103–109. [Google Scholar] [CrossRef]
- Morris, G.; Winter, J.; Cato, E.; Ritchie, A.; Bokkenheuser, V. Clostridium scindens sp. nov., a human intestinal bacterium with desmolytic activity on corticoids. Int. J. Syst. Bacteriol. 1985, 35, 478–481. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Cacciapuoti, A.F.; White, B.A.; Whitehead, T.R.; Fricke, R.J. 7α-Dehydroxylation of cholic acid by cell extracts of Eubacterium species V.P.I. 12708. Am. J. Clin. Nutr. 1980, 33, 2507–2510. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Gaskins, H.R. Another renaissance for bile acid gastrointestinal microbiology. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 348–364. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Devendran, S.; Alves, J.M.; Doden, H.; Wolf, P.G.; Pereira, G.V.; Ly, L.; Volland, A.; Takei, H.; Nittono, H. The ‘in vivo lifestyle’of bile acid 7α-dehydroxylating bacteria: Comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes 2020, 11, 381–404. [Google Scholar] [CrossRef]
- Marion, S.; Studer, N.; Desharnais, L.; Menin, L.; Escrig, S.; Meibom, A.; Hapfelmeier, S.; Bernier-Latmani, R. In vitro and in vivo characterization of Clostridium scindens bile acid transformations. Gut Microbes 2019, 10, 481–503. [Google Scholar] [CrossRef]
- Marion, S.; Desharnais, L.; Studer, N.; Dong, Y.; Notter, M.D.; Poudel, S.; Menin, L.; Janowczyk, A.; Hettich, R.L.; Hapfelmeier, S.; et al. Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res. 2020, 61, 1450–1463. [Google Scholar] [CrossRef]
- Devendran, S.; Shrestha, R.; Alves, J.M.P.; Wolf, P.G.; Ly, L.; Hernandez, A.G.; Mendez-Garcia, C.; Inboden, A.; Wiley, J.; Paul, O.; et al. Clostridium scindens ATCC 35704: Integration of nutritional requirements, the complete genome sequence, and global transcriptional responses to bile acids. Appl. Environ. Microbiol. 2019, 85, e00052-19. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.B.; Li, T.T.; Huo, D.; Qu, S.; Li, X.V.; Arifuzzaman, M.; Lima, S.F.; Shi, H.Q.; Wang, A.; Putzel, G.G.; et al. Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell 2022, 185, 547–562.e22. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Bernstein, C. Bile acids as carcinogens in the colon and at other sites in the gastrointestinal system. Exp. Biol. Med. 2023, 248, 79–89. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, W.; Chen, Y.; Huang, F.; Lu, L.; Lin, C.; Huang, T.; Ning, Z.; Zhai, L.; Zhong, L.L.; et al. A Clostridia-rich microbiota enhances bile acid excretion in diarrhea-predominant irritable bowel syndrome. J. Clin. Investig. 2020, 130, 438–450. [Google Scholar] [CrossRef]
- Ocvirk, S.; O’Keefe, S.J.D. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 2021, 73, 347–355. [Google Scholar] [CrossRef]
- Abt, M.C.; McKenney, P.T.; Pamer, E.G. Clostridium difficile colitis: Pathogenesis and host defence. Nat. Rev. Microbiol. 2016, 14, 609–620. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Osborn, L.J.; Jain, S.; Meng, X.; Weakley, A.; Yan, J.; Massey, W.J.; Varadharajan, V.; Horak, A.; Banerjee, R. Strain dropouts reveal interactions that govern the metabolic output of the gut microbiome. Cell 2023, 186, 2839–2852.e21. [Google Scholar] [CrossRef]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.L.; Cummings, B.P. The 7-α-dehydroxylation pathway: An integral component of gut bacterial bile acid metabolism and potential therapeutic target. Front. Microbiol. 2022, 13, 1093420. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Studer, N.; Desharnais, L.; Beutler, M.; Brugiroux, S.; Terrazos, M.A.; Menin, L.; Schurch, C.M.; McCoy, K.D.; Kuehne, S.A.; Minton, N.P.; et al. Functional intestinal bile acid 7α-dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model. Front. Cell Infect. Microbiol. 2016, 6, 191. [Google Scholar] [CrossRef]
- Martín, R.; Rios-Covian, D.; Huillet, E.; Auger, S.; Khazaal, S.; Bermúdez-Humarán, L.G.; Sokol, H.; Chatel, J.M.; Langella, P. Faecalibacterium: A bacterial genus with promising human health applications. FEMS Microbiol. Rev. 2023, 47, fuad039. [Google Scholar] [CrossRef]
- Bhardwaj, T.; Somvanshi, P. Pan-genome analysis of Clostridium botulinum reveals unique targets for drug development. Gene 2017, 623, 48–62. [Google Scholar] [CrossRef]
- Fernandez-Materan, F.V.; Olivos-Caicedo, K.Y.; Daniel, S.L.; Walden, K.K.; Fields, C.J.; Hernandez, A.G.; Alves, J.M.; Ridlon, J.M. Genome sequences of nine Clostridium scindens strains isolated from human feces. Microbiol. Resour. Announc. 2024, 13, e00848-24. [Google Scholar] [CrossRef] [PubMed]
- Olivos-Caicedo, K.Y.; Fernandez-Materan, F.V.; Hernandez, A.G.; Daniel, S.L.; Alves, J.M.P.; Ridlon, J.M. Complete genome sequence of the archetype bile acid 7α-dehydroxylating bacterium, Clostridium scindens VPI12708, isolated from human feces, circa 1980. Microbiol. Resour. Announc. 2023, 12, e0002923. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Patangia, D.; Almeida, A.; Zhou, Z.; Mu, D.; Paul Ross, R.; Stanton, C.; Wang, S. A compendium of 32,277 metagenome-assembled genomes and over 80 million genes from the early-life human gut microbiome. Nat. Commun. 2022, 13, 5139. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 29 January 2025).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.; Korir, D.; Ogugo, M.; Stewart, R.D.; Watson, M.; Paxton, E.; Goopy, J.; Robert, C. 1200 high-quality metagenome-assembled genomes from the rumen of African cattle and their relevance in the context of sub-optimal feeding. Genome Biol. 2020, 21, 229. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, F.B.; Maghini, D.; Oduaran, O.H.; Brewster, R.; Hulley, M.R.; Sahibdeen, V.; Norris, S.A.; Tollman, S.; Kahn, K.; Wagner, R.G.; et al. Short- and long-read metagenomics of urban and rural South African gut microbiomes reveal a transitional composition and undescribed taxa. Nat. Commun. 2022, 13, 926. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.M.; Olm, M.R.; Merrill, B.D.; Dahan, D.; Tripathi, S.; Spencer, S.P.; Feiqiao, B.Y.; Jain, S.; Neff, N.; Jha, A.R. Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell 2023, 186, 3111–3124.e13. [Google Scholar] [CrossRef] [PubMed]
- Lemos, L.N.; de Carvalho, F.M.; Santos, F.F.; Valiatti, T.B.; Corsi, D.C.; de Oliveira Silveira, A.C.; Gerber, A.; Guimarães, A.P.C.; de Oliveira Souza, C.; Brasiliense, D.M.; et al. Large scale genome-centric metagenomic data from the gut microbiome of food-producing animals and humans. Sci. Data 2022, 9, 366. [Google Scholar] [CrossRef]
- Gounot, J.S.; Chia, M.; Bertrand, D.; Saw, W.Y.; Ravikrishnan, A.; Low, A.; Ding, Y.; Ng, A.H.Q.; Tan, L.W.L.; Teo, Y.Y.; et al. Genome-centric analysis of short and long read metagenomes reveals uncharacterized microbiome diversity in Southeast Asians. Nat. Commun. 2022, 13, 6044. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef]
- Snipen, L.; Liland, K.H. micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015, 16, 79. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Wylensek, D.; Hitch, T.C.A.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernandez, S.B.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 2020, 11, 6389. [Google Scholar] [CrossRef]
- Li, T.; Yin, Y. Critical assessment of pan-genomic analysis of metagenome-assembled genomes. Brief. Bioinform. 2022, 23, bbac413. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Moldovan, M.A.; Gelfand, M.S. Pangenomic definition of prokaryotic species and the phylogenetic structure of Prochlorococcus spp. Front. Microbiol. 2018, 9, 428. [Google Scholar] [CrossRef]
- Kiu, R.; Caim, S.; Alexander, S.; Pachori, P.; Hall, L.J. Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front. Microbiol. 2017, 8, 2485. [Google Scholar] [CrossRef]
- Zou, W.; Ye, G.; Zhang, K.; Yang, H.; Yang, J. Analysis of the core genome and pangenome of Clostridium butyricum. Genome 2021, 64, 51–61. [Google Scholar] [CrossRef]
- Song, I.; Gotoh, Y.; Ogura, Y.; Hayashi, T.; Fukiya, S.; Yokota, A. Comparative genomic and physiological analysis against Clostridium scindens reveals Eubacterium sp. c-25 as an atypical deoxycholic acid producer of the human gut microbiota. Microorganisms 2021, 9, 2254. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Lee, J.W.; Cowley, E.S.; Wolf, P.G.; Doden, H.L.; Murai, T.; Caicedo, K.Y.O.; Ly, L.K.; Sun, F.; Takei, H.; Nittono, H.; et al. Formation of secondary allo-bile acids by novel enzymes from gut Firmicutes. Gut Microbes 2022, 14, 2132903. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Hylemon, P.B. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J. Lipid Res. 2012, 53, 66–76. [Google Scholar] [CrossRef]
- Harris, S.C.; Devendran, S.; Alves, J.M.P.; Mythen, S.M.; Hylemon, P.B.; Ridlon, J.M. Identification of a gene encoding a flavoprotein involved in bile acid metabolism by the human gut bacterium Clostridium scindens ATCC 35704. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 276–283. [Google Scholar] [CrossRef]
- Vital, M.; Rud, T.; Rath, S.; Pieper, D.H.; Schluter, D. Diversity of bacteria exhibiting bile acid-inducible 7α-dehydroxylation genes in the human gut. Comput. Struct. Biotechnol. J. 2019, 17, 1016–1019. [Google Scholar] [CrossRef]
- Meibom, K.L.; Marion, S.; Volet, C.; Nass, T.; Vico-Oton, E.; Menin, L.; Bernier-Latmani, R. BaiJ and BaiB are key enzymes in the chenodeoxycholic acid 7α-dehydroxylation pathway in the gut microbe Clostridium scindens ATCC 35704. Gut Microbes 2024, 16, 2323233. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Littmann, E.R.; Fontana, E.; Moody, T.U.; Kohout, C.E.; Gjonbalaj, M.; Eaton, V.; Seok, R.; Leiner, I.M.; Pamer, E.G. Functional and genomic variation between human-derived isolates of Lachnospiraceae reveals inter-and intra-species diversity. Cell Host Microbe 2020, 28, 134–146.e4. [Google Scholar] [CrossRef]
- Bai, Y.; Zhao, T.; Gao, M.; Zou, Y.; Lei, X. A novel gene alignment in Dorea sp. AM58-8 produces 7-dehydroxy-3β bile acids from primary bile acids. Biochemistry 2022, 61, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Park, D.; Jia, B.; Baek, J.H.; Hahn, Y.; Jeon, C.O. Identification and characterization of major bile acid 7α-dehydroxylating bacteria in the human gut. mSystems 2022, 7, e0045522. [Google Scholar] [CrossRef]
- Britton, G.J.; Faith, J.J. Causative microbes in host-microbiome interactions. Annu. Rev. Microbiol. 2021, 75, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.; Golan, Y.; Khanna, S.; Bobilev, D.; Erpelding, N.; Fratazzi, C.; Carini, M.; Menon, R.; Ruisi, M.; Norman, J.M.; et al. VE303, a defined bacterial consortium, for prevention of recurrent Clostridioides difficile infection: A randomized clinical trial. JAMA 2023, 329, 1356–1366. [Google Scholar] [CrossRef]
- Oliveira, R.A.; Pamer, E.G. Assembling symbiotic bacterial species into live therapeutic consortia that reconstitute microbiome functions. Cell Host Microbe 2023, 31, 472–484. [Google Scholar] [CrossRef]
- Dsouza, M.; Menon, R.; Crossette, E.; Bhattarai, S.K.; Schneider, J.; Kim, Y.G.; Reddy, S.; Caballero, S.; Felix, C.; Cornacchione, L.; et al. Colonization of the live biotherapeutic product VE303 and modulation of the microbiota and metabolites in healthy volunteers. Cell Host Microbe 2022, 30, 583–598.e8. [Google Scholar] [CrossRef]
- Mohanty, I.; Allaband, C.; Mannochio-Russo, H.; El Abiead, Y.; Hagey, L.R.; Knight, R.; Dorrestein, P.C. The changing metabolic landscape of bile acids—Keys to metabolism and immune regulation. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 493–516. [Google Scholar] [CrossRef]
- Wang, S.; Xu, C.; Liu, H.; Wei, W.; Zhou, X.; Qian, H.; Zhou, L.; Zhang, H.; Wu, L.; Zhu, C.; et al. Connecting the gut microbiota and neurodegenerative diseases: The role of bile acids. Mol. Neurobiol. 2023, 60, 4618–4640. [Google Scholar] [CrossRef]
- Callender, C.; Attaye, I.; Nieuwdorp, M. The interaction between the gut microbiome and bile acids in cardiometabolic diseases. Metabolites 2022, 12, 65. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Trauner, M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 432–450. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Contigs or Scaffolds (Genome Assembly Level) | Accession/RefSeq Number | Host (Source) | Geographic Origin | BioSample | BioProject | Genome Assembly Identifier |
|---|---|---|---|---|---|---|---|
| JCM10419 | 1 (not circularized) | CP137824 | Homo sapiens (feces) | Japan | SAMN37482747 | PRJNA1026650 | ASM3353943v1 |
| JCM10420 | 1 (closed) | CP137823 | Homo sapiens (feces) | Japan | SAMN37482748 | PRJNA1026650 | ASM3353941v1 |
| JCM10422 | 2 contigs | CP137821-CP137822 | Homo sapiens (feces) | Japan | SAMN37482749 | PRJNA1026650 | ASM4093202v1 |
| JCM10423 | 1 (not circularized) | CP137820 | Homo sapiens (feces) | Japan | SAMN37482750 | PRJNA1026650 | ASM3353951v1 |
| I10 | 1 (closed) | CP137819 | Homo sapiens (feces) | Japan | SAMN37482751 | PRJNA1026650 | ASM3353949v1 |
| MO32 | 1 (closed) | CP137818 | Homo sapiens (feces) | Japan | SAMN37482752 | PRJNA1026650 | ASM3353945v1 |
| NT182 | 1 (closed) | CP137817 | Homo sapiens (feces) | Japan | SAMN37482753 | PRJNA1026650 | ASM3353953v1 |
| S076 | 1 (closed) | CP137816 | Homo sapiens (feces) | Japan | SAMN37482754 | PRJNA1026650 | ASM3353947v1 |
| S077 | 5 contigs | CP137811-CP137815 | Homo sapiens (feces) | Japan | SAMN37482755 | PRJNA1026650 | ASM4093201v1 |
| VPI12708 | 1 (closed) | CP113781 | Homo sapiens (feces) | Germany | SAMN31775693 | PRJNA902789 | ASM2794165v1 |
| CE91—St59 | 1 (closed) | AP025569.1 | Homo sapiens (feces) | Japan | SAMD00389867 | PRJDB11902 | ASM2284581v1 |
| CE91—St60 | 1 (closed) | AP025570.1 | Homo sapiens (feces) | Japan | SAMD00389868 | PRJDB11902 | ASM2284583v1 |
| G10 | 1 (closed) | AP024846.1 | Rattus norvegicus (cecal content) | Japan | SAMD00239677 | PRJDB10323 | ASM2089211v1 |
| Q4 | 1 (closed) | CP080442.1 | Homo sapiens (feces) | USA | SAMN20488193 | PRJNA750754 | ASM1959792v1 |
| BL389WT3D | 1 (closed) | CP045695.1 | Sus scrofa domesticus (feces) | Germany | SAMN13152203 | PRJNA561470 | ASM968469v1 |
| FDAARGOS_1227 | 1 (closed) | CP069444.1 | Not available | USA | SAMN16357369 | PRJNA231221 | ASM1688900v1 |
| ATCC 35704 | 1 (closed) | CP036170.1 | Homo sapiens (feces) | USA | SAMN10519000 | PRJNA508260 | ASM429512v1 |
| AM05-22 | 56 scaffolds | GCF_027662895.1 | Homo sapiens (feces) | China | SAMN31808509 | PRJNA903559 | ASM2766289v1 |
| AM07-30 | 50 scaffolds | GCF_027662765.1 | Homo sapiens (feces) | China | SAMN31808516 | PRJNA903559 | ASM2766276v1 |
| SL.1.22 | 52 contigs | GCF_020555615.1 | Homo sapiens (feces) | USA | SAMN22167568 | PRJNA737800 | ASM2055561v1 |
| DFI.1.234 | 107 contigs | GCF_022137935.1 | Homo sapiens (feces) | USA | SAMN24725968 | PRJNA792599 | Not available |
| GGCC_0168 | 256 contigs | GCF_017565985.1 | Homo sapiens (feces) | USA | SAMN14737934 | PRJNA628657 | ASM1756598v1 |
| DFI.1.217 | 96 contigs | GCF_020562885.1 | Homo sapiens (feces) | USA | SAMN22167352 | PRJNA737800 | ASM2056288v1 |
| DFI.1.162 | 125 contigs | GCF_020563365.1 | Homo sapiens (feces) | USA | SAMN22167324 | PRJNA737800 | ASM2056336v1 |
| DFI.1.161 | 160 contigs | GCF_024463895.1 | Homo sapiens (feces) | USA | SAMN28944463 | PRJNA792599 | ASM2446389v1 |
| MSK.1.26 | 93 contigs | GCF_013304105.1 | Homo sapiens (feces) | USA | SAMN14067588 | PRJNA596270 | ASM1330410v1 |
| DFI.1.60 | 197 contigs | GCF_020561885.1 | Homo sapiens (feces) | USA | SAMN22167389 | PRJNA737800 | ASM2056188v1 |
| MSK.1.16 | 93 contigs | GCF_013304115.1 | Homo sapiens (feces) | USA | SAMN14067587 | PRJNA596270 | ASM2056188v1 |
| MSK.5.24 | 21 contigs | GCF_013304085.1 | Homo sapiens (feces) | USA | SAMN14067589 | PRJNA596270 | ASM1330408v1 |
| DFI.1.130 | 797 contigs | GCF_020563525.1 | Homo sapiens (feces) | USA | SAMN22167316 | PRJNA737800 | ASM2056352v1 |
| DFI.4.63 | 195 contigs | GCF_020560435.1 | Homo sapiens (feces) | USA | SAMN22167449 | PRJNA737800 | ASM2056043v1 |
| MGYG-HGUT-01303 | 41 scaffolds | GCF_902373645.1 | Homo sapiens (feces) | Not available | SAMEA5850806 | PRJEB33885 | MGYG-HGUT-01303 |
| NB2A-7-D5 | 39 contigs | GCF_024125195.1 | Homo sapiens (feces) | Not available | SAMN28102059 | PRJNA835435 | ASM2412519v1 |
| VE202-05 | 102 contigs | Not available | Homo sapiens (feces) | Japan | SAMD00004073 | PRJDB524 | ASM47184v1 |
| Strain | G + C% | tmRNA | tRNA | rRNA | CDS | Genome Size (bp) | No. Contigs | |
|---|---|---|---|---|---|---|---|---|
| 1 | I10 | 46.5 | 2 | 56 | 12 | 3602 | 3,435,295 | 1 |
| 2 | JCM10419 | 47.5 | 1 | 57 | 12 | 3682 | 3,940,699 | 1 |
| 3 | JCM10420 | 47.4 | 1 | 57 | 11 | 3768 | 4,020,045 | 1 |
| 4 | JCM10422 | 46.6 | 1 | 56 | 12 | 3390 | 3,501,106 | 2 |
| 5 | JCM10423 | 46.9 | 1 | 57 | 11 | 4057 | 4,306,053 | 1 |
| 6 | MO32 | 47.8 | 1 | 57 | 11 | 3691 | 3,929,075 | 1 |
| 7 | NTI82 | 47.1 | 1 | 59 | 11 | 4137 | 4,318,168 | 1 |
| 8 | S076 | 47.3 | 1 | 58 | 11 | 4103 | 4,290,604 | 1 |
| 9 | S077 | 46.5 | 2 | 57 | 12 | 3386 | 3,403,497 | 5 |
| 10 | VPI 12708 * | 47.7 | 1 | 56 | 12 | 3716 | 3,983,052 | 1 |
| 11 | ATCC 35704 * | 46.3 | 2 | 58 | 12 | 3656 | 3,658,040 | 1 |
| 12 | BL389WT3D * | 45.5 | 2 | 58 | 12 | 3655 | 3,785,527 | 1 |
| 13 | CE91-St59 * | 45.9 | 2 | 56 | 12 | 3602 | 3,608,085 | 1 |
| 14 | CE91-St60 * | 45.9 | 2 | 56 | 12 | 3608 | 3,608,087 | 1 |
| 15 | FDAARGOS_1227 * | 46.3 | 2 | 57 | 12 | 3603 | 3,619,096 | 1 |
| 16 | G10 * | 46.6 | 2 | 57 | 12 | 3295 | 3,315,593 | 1 |
| 17 | Q4 * | 47.7 | 1 | 57 | 12 | 3725 | 3,941,835 | 1 |
| 18 | AM05-22 * | 46.5 | 2 | 54 | 3 | 3269 | 3,330,149 | 56 |
| 19 | AM07-30 * | 46.5 | 2 | 49 | 2 | 3272 | 3,331,670 | 50 |
| 20 | DFI.1.130 * | 46.9 | 1 | 53 | 4 | 4177 | 4,565,863 | 797 |
| 21 | DFI.1.161 * | 47.1 | 1 | 51 | 4 | 4041 | 4,325,545 | 160 |
| 22 | DFI.1.162 * | 46.9 | 1 | 52 | 4 | 4161 | 4,396,306 | 125 |
| 23 | DFI.1.217 * | 46.9 | 1 | 52 | 4 | 4161 | 4,389,871 | 96 |
| 24 | DFI.1.234 * | 47 | 1 | 52 | 4 | 4072 | 4,316,625 | 107 |
| 25 | DFI.1.60 * | 47.1 | 1 | 52 | 4 | 4047 | 4,309,437 | 197 |
| 26 | DFI.4.63 * | 47.5 | 1 | 52 | 4 | 3958 | 4,167,375 | 195 |
| 27 | GGCC_0168 * | 46.6 | 2 | 57 | 12 | 3390 | 3,417,088 | 256 |
| 28 | MGYG-HGUT-01303 * | 46.4 | 2 | 64 | 22 | 3574 | 3,622,605 | 41 |
| 29 | MSK.1.16 * | 46.4 | 2 | 54 | 4 | 3143 | 3,230,100 | 93 |
| 30 | MSK.1.26 * | 46.4 | 2 | 54 | 3 | 3140 | 3,227,969 | 93 |
| 31 | MSK.5.24 * | 47.5 | 1 | 54 | 4 | 3845 | 4,072,709 | 21 |
| 32 | NB2A-7-D5 * | 47.5 | 1 | 60 | 4 | 3980 | 4,182,602 | 39 |
| 33 | SL.1.22 * | 47.5 | 1 | 55 | 3 | 3706 | 3,970,092 | 52 |
| 34 | VE202-05 * | 47.8 | 0 | 48 | 2 | 4523 | 3,912,387 | 102 |
| Completeness of MAGs % | Pangenome | Core Genome |
|---|---|---|
| ≥95 | 14,625 | 850 |
| ≥90 | 17,713 | 401 |
| ≥85 | 19,189 | 132 |
| ≥80 | 19,792 | 75 |
| ≥75 | 21,923 | 8 |
| ≥70 | 22,864 | 4 |
| Completeness of MAGs % | Pangenome | Core Genome |
|---|---|---|
| ≥95 | 14,464 | 1120 |
| ≥90 | 15,144 | 931 |
| ≥85 | 16,168 | 501 |
| ≥80 | 16,629 | 276 |
| ≥75 | 19,083 | 25 |
| ≥70 | 20,023 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivos-Caicedo, K.Y.; Fernandez-Materan, F.V.; Daniel, S.L.; Anantharaman, K.; Ridlon, J.M.; Alves, J.M.P. Pangenome Analysis of Clostridium scindens: A Collection of Diverse Bile Acid- and Steroid-Metabolizing Commensal Gut Bacterial Strains. Microorganisms 2025, 13, 857. https://doi.org/10.3390/microorganisms13040857
Olivos-Caicedo KY, Fernandez-Materan FV, Daniel SL, Anantharaman K, Ridlon JM, Alves JMP. Pangenome Analysis of Clostridium scindens: A Collection of Diverse Bile Acid- and Steroid-Metabolizing Commensal Gut Bacterial Strains. Microorganisms. 2025; 13(4):857. https://doi.org/10.3390/microorganisms13040857
Chicago/Turabian StyleOlivos-Caicedo, Kelly Y., Francelys V. Fernandez-Materan, Steven L. Daniel, Karthik Anantharaman, Jason M. Ridlon, and João M. P. Alves. 2025. "Pangenome Analysis of Clostridium scindens: A Collection of Diverse Bile Acid- and Steroid-Metabolizing Commensal Gut Bacterial Strains" Microorganisms 13, no. 4: 857. https://doi.org/10.3390/microorganisms13040857
APA StyleOlivos-Caicedo, K. Y., Fernandez-Materan, F. V., Daniel, S. L., Anantharaman, K., Ridlon, J. M., & Alves, J. M. P. (2025). Pangenome Analysis of Clostridium scindens: A Collection of Diverse Bile Acid- and Steroid-Metabolizing Commensal Gut Bacterial Strains. Microorganisms, 13(4), 857. https://doi.org/10.3390/microorganisms13040857