Abstract
Antibiotic resistance to Klebsiella pneumoniae poses a major public health threat, particularly in intensive care unit (ICU) settings. The emergence of extensively drug-resistant (XDR) strains complicates treatment options, requiring a deeper understanding of their genetic makeup and potential therapeutic targets. This research delineated an extensively drug-resistant (XDR) Klebsiella pneumoniae strain obtained from an ICU patient and telomeric temperate phage derived from hospital effluent. The bacteria showed strong resistance to multiple antibiotics, including penicillin (≥16 μg/mL), ceftriaxone (≥32 μg/mL), and meropenem (≥8 μg/mL), which was caused by SHV-11 beta-lactamase, NDM-1 carbapenemase, and porin mutations (OmpK37, MdtQ). The strain was categorized as K46 and O2a types and carried virulence genes involved in iron acquisition, adhesion, and immune evasion, as well as plasmids (IncHI1B_1_pNDM-MAR, IncFIB) and eleven prophage regions, reflecting its genetic adaptability and resistance dissemination. The 172,025 bp linear genome and 46.3% GC content of the N-15-like phage showed strong genomic similarities to phages of the Sugarlandvirus genus, especially those that infect K. pneumoniae. There were structural proteins (11.8%), DNA replication and repair enzymes (9.3%), and a toxin–antitoxin system (0.4%) encoded by the phage genome. A protelomerase and ParA/B partitioning proteins indicate that the phage is replicating and maintaining itself in a manner similar to the N15 phage, which is renowned for maintaining a linear plasmid prophage throughout lysogeny. Understanding the dynamics of antibiotic resistance and pathogen development requires knowledge of phages like this one, which are known for their temperate nature and their function in altering bacterial virulence and resistance profiles. The regulatory and structural proteins of the phage also provide a model for research into the biology of temperate phages and their effects on microbial communities. The importance of temperate phages in bacterial genomes and their function in the larger framework of microbial ecology and evolution is emphasized in this research.
1. Introduction
The development of extensively drug-resistant (XDR) bacterial pathogens poses a major threat to public health worldwide. Now, a major pathogen causing hospital-acquired infections, including urinary tract infections, pneumonia, and bloodstream infections, is Klebsiella pneumoniae [1]. The reputation of K. pneumoniae as a major nosocomial pathogen has been much boosted by its ability to acquire and disseminate antibiotic resistance genes via horizontal gene transfer routes, including plasmids, transposons, and prophages [2]. Antibiotic resistance genes (ARGs), especially those that code for beta-lactamases and carbapenemases, have made many first-line antibiotics less effective [3]. This makes treatment plans more difficult and puts more stress on healthcare systems [4]. Virulence factors, including siderophores (e.g., yersiniabactin and enterobactin) and adhesion systems (e.g., E. coli common pilus), increase the bacterium’s potential to colonize host tissues, avoid immune responses, and start chronic infections [5]. Developing sensible plans to treat K. pneumoniae depends on an understanding of the genetic pathways leading to antibiotic resistance and virulence [6]. By means of horizontal gene transfer (HGT) and manipulation of bacterial virulence and resistance traits, bacteriophages, also known as phages, affect bacterial evolution to a large degree [7]. Temperate phages integrate with bacterial genomes as prophages and may contain genes that provide their hosts with selection benefits, such as antibiotic resistance or virulence factors [8]. The linear genome of the N15-like phage distinguishes it from most other temperate phages that do not merge into the host chromosome; it survives as a linear plasmid during the lysogenic stage [9,10]. Protelomerase, which changes the linear phage genome into a circular form during replication, and partitioning proteins, which help the phage genome to be stably segregated during cell division, highlight several mechanisms the phage uses to preserve its genetic material, helping the phage to survive among bacterial populations and increasing their genetic variation [11]. This replication system guarantees consistent phage genome inheritance and helps bacterial hosts horizontally transmit genetic material, including ARGs and virulence factors [11]. Researching these phages helps us grasp the dynamics of host–phage interactions and the processes behind phage-mediated bacterial evolution [8,9]. The phage genome codes for a lot of different proteins, such as structural proteins, DNA replication and repair enzymes, and toxin–antitoxin systems that make it easier for the genome to infect, multiply, and stay inside bacterial hosts [12]. Analyzing temperate phages helps explain bacterial evolution, host–phage interactions, and antibiotic resistance [13]. Phages increase bacterial pathogen genetic diversity and adaptation, which is crucial to understanding antibiotic resistance and pathogen development [8]. The phage’s structural and regulatory proteins mimic temperate phage biology and their effects on microbial communities [14].
Although lytic phages are favored by therapeutics because of their rapid bacterial elimination, temperate phages are promising candidates for therapy and phage engineering because of their ability to integrate into bacterial genomes and carry useful genes [7,15]. Temperate phages may persist in bacteria and disseminate genetic material that can kill bacteria or reduce bacterial virulence or antibiotic resistance [16]. The engineering of therapeutic temperate phages has improved with synthetic biology where temperate phages can acquire CRISPR-Cas technology to delete antibiotic resistance or virulence genes [17,18]. Temperate phages may delete the integration mechanism to become “lytic-only” phages that infect and lyse bacteria [19]. Engineering projects take advantage of the phage linear plasmid (like telomeric phages) in phage therapy and for the expression of recombination proteins [20]. The present investigation examines an N15-like phage identified in hospital wastewater and an extensively drug-resistant K. pneumoniae strain obtained from a clinical environment. The genetic and functional traits of the bacterial strain and the phage investigated in this study provide valuable insights into the interactions between bacteria and phages, as well as their implications for pathogen evolution and antibiotic resistance. This study underscores the significance of temperate phages in bacterial genomes and their contributions to microbial ecology and evolution, thereby indicating the necessity for ongoing research into phage–bacteria interactions to combat antibiotic-resistant diseases.
2. Material and Methods
2.1. Culture Conditions and Bacterial Strains
Intensive care units (ICUs) extensively drug-resistant (XDR) clinical strain of K. pneumoniae was isolated from sputum sample. Bacterial identification and the antibiotic susceptibility profiles and MICs of the isolates were assessed by the VITEK 2 system (VITEK® 2, BIOMÉRIEUX, Craponne, France). Bacterial strains were cultivated in Luria–Bertani (LB) broth or on LB agar plates at 37 °C and preserved at −80 °C in glycerol stocks for long-term storage.
2.2. Bacteriophage Isolation and Purification
Environmental samples (hospital effluents) were collected in a sterile 1 L sterile glass bottle for phage isolation. The enrichment method was used, where 10 mL of sample was mixed with 10 mL of LB broth containing a bacterial host, incubated at 37 °C for 24 h with shaking, and filtered through a 0.22 µm membrane [21]. The resulting phage suspension was serially diluted and plated on soft LB agar containing the host bacteria to detect plaque formation. Single plaques were picked, purified by repeated plaque assays, and stored at 4 °C.
2.3. Phage Characterization
Phage morphology was observed using transmission electron microscopy (TEM) according to standard procedures [22]. Briefly, a drop of the purified phage suspension was placed on a carbon-coated copper grid, stained with 2% uranyl acetate, and examined using a JEOL JEM-1400 electron microscope (Tokyo, Japan) at 120 kV.
The host range of the isolated phage was determined by performing plaque assays against a panel of clinical K. pneumoniae strains (from our lab stock) and E. coli ATCC 25922™, K. pneumoniae ATCC13883™ and P. aeruginosa ATCC 9027™. The strains were cultured overnight and mixed with soft agar containing the host bacterial strains, and plaque formation was observed to determine the host range of the phage [23].
2.4. Bacterial DNA Isolation, Sequencing and Bioinformatics Analysis
The bacterial genomic DNA of K. pneumoniae ST147 Kpn-R1 was isolated using the Qiagen Blood and Tissue DNA Extraction Kit, following the manufacturer’s protocol. DNA purity and concentration were verified using a nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Whole-genome sequencing (WGS) was performed by Macrogen, Seoul, Republic of Korea on the Illumina NovaSeq 6000 platform (2 × 150 bp paired-end reads) (Illumina, San Diego, CA, USA). Libraries were prepared using the TruSeq Nano DNA Kit (350 bp insert size) (Illumina, San Diego, CA, USA), with library quality assessed via the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and TapeStation D1000 Screen Tape (Agilent Technologies, Santa Clara, CA, USA). Raw sequencing reads were trimmed with Trimmomatic (v0.39) to remove adapters and low-quality bases (parameters: ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10, LEADING:20, TRAILING:20, SLIDINGWINDOW:4:20, MINLEN:50) [24]. The cleaned reads were assembled into contigs using SPAdes (v3.15.5) with default settings [25]. Raw data from whole-genome sequencing (WGS) were integrated using Proksee Assemble (v1.3.0) [26]. Genome annotation was performed using Prokka (v1.2.0), which identified open reading frames (ORFs), tRNA, rRNA, and other genomic features [27]. Map Builder (v2.0.5) generated a CGView JSON file for showing the completed genome from GenBank formats. To rank BLAST tracks based on similarity and color BLAST features based on percent identity, the BLAST Formatter (v1.0.3) was used, assisting in the identification of conserved and divergent regions.
Alien Hunter (v1.1.0) was used to forecast likely horizontal gene transfer (HGT) events based on atypical nucleotide compositions that indicate alien DNA acquisition [28]. To compare the genome with closely related species and determine taxonomic connections, the whole-genome average nucleotide identity (ANI) was calculated using FastANI (v1.1.0) [29]. The Comprehensive Antibiotic Resistance Database (CARD) Resistance Gene Identifier (RGI) (v1.2.1) was used to identify antibiotic resistance genes. Based on the CARD database, this tool anticipates resistance genes and mutations, giving information on the genome’s antibiotic resistance profile [30]. VirSorter (v1.1.1) was used to find viral genomes [31]. Phigaro (v1.0.1) has been annotated in prophage regions [32]. Mobile OG-db (v1.1.3), a database and tool designed for the identification of mobile genetic elements (MGEs), including plasmids, transposons, and integrons [33]. The CRISPR/Cas Finder (v1.1.0) tool was used to locate CRISPR arrays and associated Cas proteins, offering insights into the genome’s adaptive immune system [34]. The Proksee tool displayed and organized all genetic traits. All visible tracks on the genome map were documented in a detailed figure caption created using the Track List Caption (v1.2.0) [26].
Using Kaptive Web [35], capsule (K) and lipopolysaccharide (O) serotypes in K. pneumoniae genomes were predicted. Sequence-based typing and known Klebsiella strain comparisons were made using the PubMLST Klebsiella database [36]. Virulence factors were identified using the 2019 Virulence Factor Database (VFDB) [37]. Barrnap helped pull out the 16S rRNA gene sequence [38]. By use of NCBI BLASTn database comparison, closely similar species were identified. A phylogenetic tree based on the 16S rRNA sequence and other conserved markers was built using MEGA 12 [39], therefore exposing the evolutionary links of the isolate, with the final genome deposited in GenBank under BioProject PRJNA1217456.
2.5. Phage DNA Isolation, Sequencing, and Bioinformatics Analysis
Phage DNA was extracted from purified lysates using the Norgen Biotek Phage DNA Isolation Kit (Norgen Biotek Corp., Thorold, ON, Canada). Sequencing libraries were prepared identically to bacterial methods (TruSeq Nano DNA Kit, Illumina, San Diego, CA, USA; Illumina NovaSeq 6000, Illumina, San Diego, CA, USA). Raw reads were processed with Trimmomatic (same parameters as above) and assembled using SPAdes (v3.15.5). Additionally, the quality of the genome assembly was checked using FastQC (v0.12.1) and QUAST (v5.2.0) [40]. Genome completeness was ascertained using CheckV (v1.0.1) [41], and structural features were projected using PhageTerm (v4.0.0) [42]. Functional annotations included phage lifestyle prediction using Bacphlip (v0.9.6) [43] and protein classification using PhANNs [44]. Comparative genomics was run using Pyani (v0.2.12) [45] and Clinker (v0.0.24) [46]. AMRFinderPlus (v3.12.8) [47] and VirulenceFinder (v2.0.4) [48] separately verified antimicrobial resistance and virulence genes. The phage genome was deposited in GenBank under accession numbers PQ800144.1–PQ800159.1.
2.6. Statistical Analysis
Statistical analyses were conducted using R (v4.3.1) and Python (v3.10) to evaluate genomic data. Descriptive statistics summarized key metrics (e.g., genome size, GC content, gene coverage). Comparative genomic analyses included average nucleotide identity (ANI) calculations using FastANI and MASH distances for phylogenetic comparisons. Multiple testing corrections (Benjamini–Hochberg method, FDR < 0.05) were applied where applicable. For phylogenetic analyses, branch support was assessed via 500 bootstrap replicates in MEGA12. Correlation analyses between genetic features (e.g., plasmid replicons, prophage regions) and antibiotic resistance profiles used logistic regression models (R stats package, v4.4.3, R Core Team, 2025). MASH distances were used to show the genome in CGView (v1.1.1) [49] against the Millardlab Phage Database [50]. Phylogenetic trees and genomic similarity matrices were computed using Viptree and Clinker for synteny analysis [51].
3. Results
3.1. Genome Assembly and Strain Phylogenetic Analysis
The total genome assembly of the strain was 5,814,374 bp with a GC content of 56.84%. The assembly was of high quality, with 0.00 N’s per 100 kbp, indicating no ambiguous bases in the 175 contigs. These metrics collectively demonstrate a robust and continuous genome assembly, suitable for downstream analysis. MLST analysis revealed that the strain belongs to the ST147 high-risk clone according to the Pasteur database.
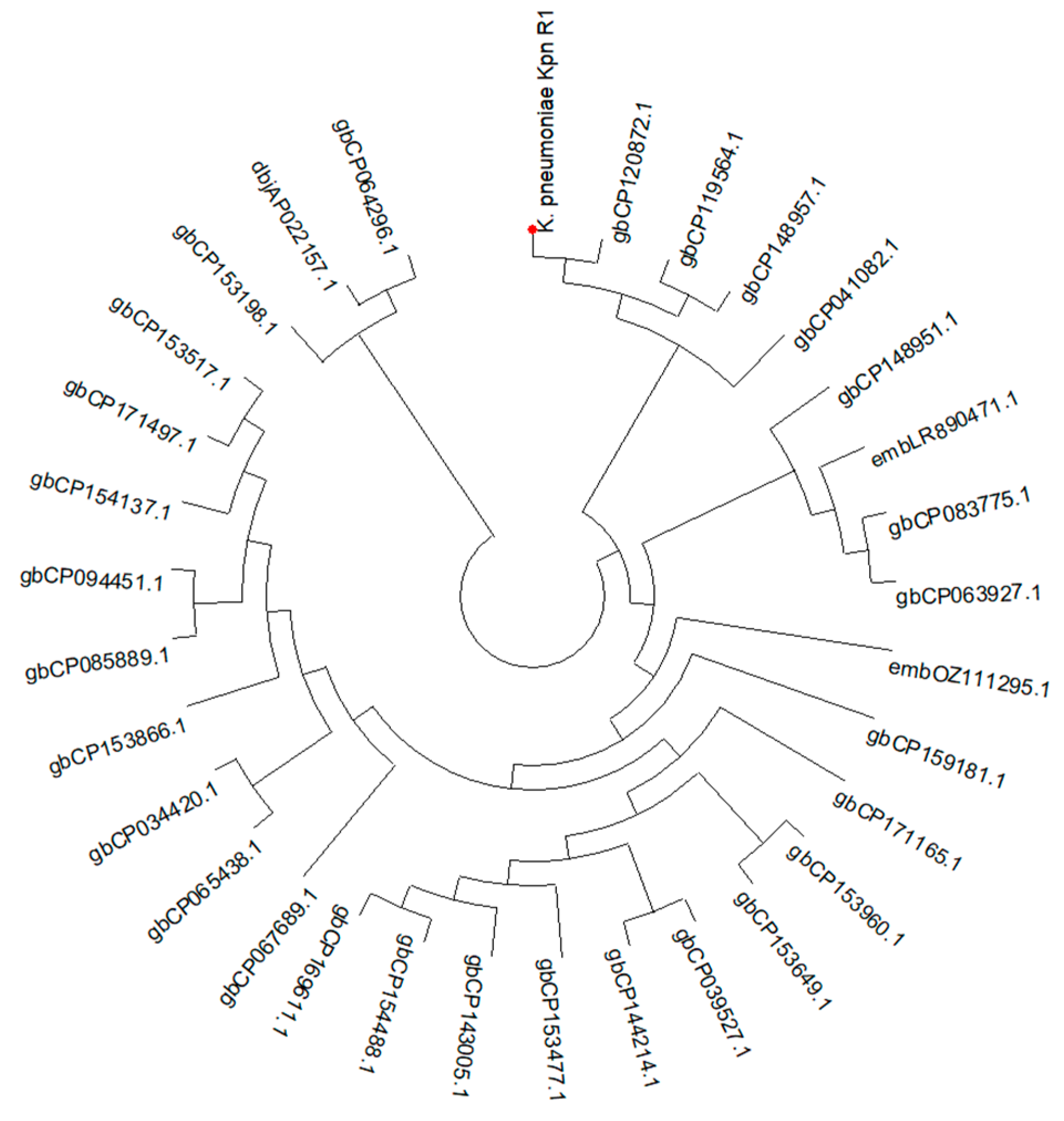
The phylogenetic investigation (Figure 1) indicates a close evolutionary link between K. pneumoniae kpn_R01 and the clinical isolate CP120872 from China. Clinical isolates CP148957 from Japan and CP119564 from the United Kingdom further cluster with this strain, suggesting potential global transmission. The extended family tree includes CP063927, a Chinese environmental isolation, and CP083775, an Australian clinical isolate. The proximity of environmental and clinical isolates indicates that environmental reservoirs contributed to the transmission of the lineage. The identification of analogous strains in China, Japan, the United Kingdom, and Australia contributes to the evidence of potential global dissemination. This may result from infections acquired during hospital care or while patients are abroad.
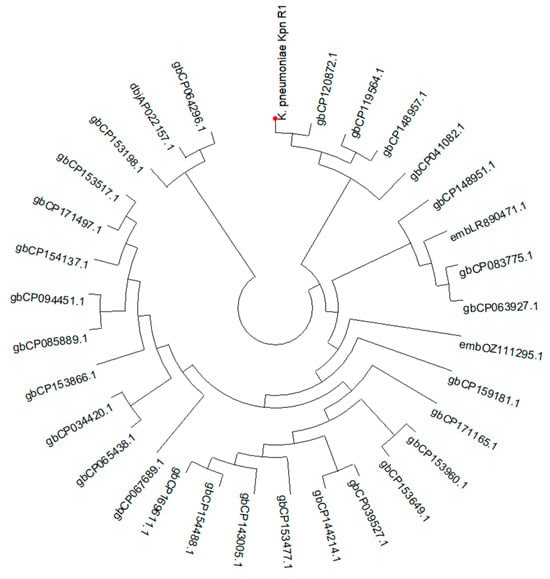
Figure 1.
A circular phylogenetic tree illustrating the evolutionary relationships of K. pneumoniae Kpn_R01 and its 34 closest strains. The tree was inferred from 16S rRNA gene sequences using the maximum likelihood (ML) tree highlighting the phylogenetic placement of Kpn_R01 (marked with a red dot) among closely related strains. Clustering patterns indicate potential global dissemination, with strains originating from diverse geographic locations. The close association between clinical and environmental isolates underscores the role of environmental reservoirs in bacterial transmission. The tree was inferred using the ML method with the Tamura–Nei model [52], and branch support was assessed with 500 bootstrap replicates [53]. The initial heuristic search compared a neighbor-joining (NJ) tree [54] and a maximum parsimony (MP) tree, selecting the one with the best log-likelihood score. Evolutionary analyses were conducted in MEGA12 [55].
3.2. Antimicrobial Susceptibility and Resistome
K. pneumoniae Kpn_R01 is categorized as extensively drug-resistant (XDR) owing to its resistance to multiple antibiotic classes. This classification is supported by minimum inhibitory concentration (MIC) testing, as presented in Table 1, and is further validated through resistance gene analysis utilizing the CARD database. Extensively drug-resistant (XDR) bacteria are defined globally as exhibiting resistance to at least one agent in all but one or two categories of antimicrobials [56]. This strain qualifies for XDR designation as it exhibits resistance to beta-lactams, cephalosporins, carbapenems, monobactams, fluoroquinolones, aminoglycosides, tetracyclines, macrolides, polymyxins, sulfonamides, trimethoprim, phenicols, rifamycins, and fosfomycin. This strain demonstrates resistance to a wide array of antibiotics; however, additional research is required to ascertain whether drugs continue to be effective in addressing pandrug resistance (PDR) criteria. As shown in Table 1, SHV-11 beta-lactamase (blaSHV-11), NDM-1 carbapenemase (blaNDM-1), and mutations in porins, encoded by OmpK37 and MdtQ, mediate the strain resistance to beta-lactam antibiotics, resulting in minimum inhibitory concentrations (MICs) of penicillin (≥16 μg/mL), ceftriaxone (≥32 μg/mL), and meropenem (≥8 μg/mL). Aztreonam resistance (≥32 μg/mL) is attributed to low permeability caused by outer membrane porin mutations (OmpK37) and efflux (MdtQ). Minimum inhibitory concentrations (MICs) of ciprofloxacin (≥4 μg/mL) and levofloxacin (≥8 μg/mL) are primarily due to fluoroquinolones being augmented by efflux pumps (oqxA, acrA, marA) and target mutations (rsmA). Enhanced aminoglycoside resistance is ascribed to 16S rRNA methyltransferase (armA) and aminoglycoside-modifying enzymes (aadA2), leading to minimum inhibitory concentrations (MICs) of ≥16 μg/mL for gentamicin and ≥64 μg/mL for amikacin. Resistance to tetracycline (≥16 μg/mL) and tigecycline (≥2 μg/mL) is facilitated by efflux pumps (tet(A) and KpnF). Efflux pumps (KpnF, KpnG, KpnH) and enzymatic inactivation (msrE and mphE) promote resistance to macrolide, resulting in minimum inhibitory concentrations (MICs) of ≥8 μg/mL for erythromycin and ≥16 μg/mL for azithromycin. Resistance to colistin and polymyxin B (≥4 μg/mL) is associated with phosphoethanolamine transferase (ArnT and eptB) and reduced permeability (OmpA). Increased resistance to sulfamethoxazole (≥256 μg/mL) and trimethoprim (≥16 μg/mL) is ascribed to target replacement (sul1 and dfrA12). Fosfomycin resistance (≥64 μg/mL) arises from enzymatic inactivation (fosA5). Chloramphenicols, rifamycins, and quaternary ammonium compound resistance is primarily due to the efflux pump. The extensive resistance profile of this strain highlights the challenges in managing infections caused by XDR K. pneumoniae and underscores the need for alternative treatment strategies.

Table 1.
The minimum inhibitory concentrations (MICs) for various antimicrobials and the resistance mechanisms identified in the strain.
3.3. Genes Linked to Virulence
Table 2 shows that K. pneumoniae Kpn_R01 strain harbors a number of important virulence factors that were identified via the examination of virulence-associated genes. These factors included genes linked to the production of yersiniabactin, enterobactin, and the E. coli common pilus (ECP). High coverage (98.31% to 100%) and identity (85.28% to 90.07%) of the ECP gene cluster (ecpR, ecpA, ecpB, ecpC, ecpD, and ecpE) indicates that the strain may manufacture E. coli common pili, which are important in adhesion and biofilm formation. This strain has the ability to produce yersiniabactin, a siderophore that aids in iron acquisition and virulence; the yersiniabactin biosynthetic gene cluster (fyuA, ybtE, ybtT, ybtU, irp1, irp2, ybtA, ybtP, ybtQ, ybtX, and ybtS) was also detected with 100% coverage and 97.62% to 99.95% identity. The strain has the ability to produce enterobactin, a siderophore that plays a crucial role in iron absorption. Genes involved in its production were identified (fepC, fepG, entB, and entA), and the coverage and identity were (88.72% to 99.33%) and (80.00% to 82.65%), respectively. In addition to being discovered with 100% coverage and 83.75% identity, the ompA gene codes for the outer membrane protein A; hence, this strain may be able to elude the immune system and cause diseases. Among these outcomes are the strain’s pathogenicity-critical capabilities—its ability to adhere, absorb iron, and evade the immune system.
Table 2.
Virulence-associated genes identified in the strain.
3.4. K- and O Serotypes
According to the results of the Kaptive analysis shown in Table S1, there are two distinct loci that correspond to the K64 and O2a serotypes, respectively: KL64 and O1/O2v1.
With 100% identity and 90.10% coverage, indicating a partial match, the KL64 locus was determined to be typeable. Out of the 24 anticipated genes in the KL64 locus, 23 were identified (95.83%), with 1 gene (KL64_12_wcoT) absent. Numerous genes within the KL64 locus were either partly shortened or incomplete, including wzx, wcoV, wzy, wcoU, and wcsF, potentially impacting the functioning of the capsular polysaccharide production pathway. Furthermore, three other genes outside the locus were found, namely, rfaG, gmd, and HG290, which are linked to other serotypes (KL40, KL150, and KL4).
The O1/O2v1 locus indicated that the strain was accurately categorized as O2a, exhibiting 99.03% identity and 100% coverage. The seven O-locus genes (wzm, wzt, wbbM, glf, wbbN, wbbO, and kfoC) were predicted as present and undamaged with high identity and coverage ranging from 97.31% to 100%. In addition to the locus, six other genes were identified: manC, manB, rfbB, rfbD, rfbA, and rfbC. These genes are often associated with different serotypes, such as O3b, O12, and OL13. These results indicate a highly preserved O2a serotype with possible genetic interchange with other serotypes. The strain exhibits a well-preserved O2a serotype along with supplementary genetic components that may enhance its antigenic variety and indicate a possible genetic exchange with other serotypes.
3.5. Plasmids
Several plasmid replicons and colicin genes were found in the strain’s examination of plasmid-associated genes, suggesting that the plasmid content is varied (Table 3). A plasmid linked to NDM carbapenemase resistance may be present in the IncHI1B_1_pNDM-MAR replicon, which was found with 100% coverage and identity. Also present were IncFIB (K)_1_Kpn3 and IncFIB (pKPHS1)_1_pKPHS1 replicons, which are often associated with K. pneumoniae, with 100% coverage and 95.54% and 91.07% identity, respectively. The presence of high coverage (100%) and identity (88.60% to 100%) for colicin genes, such as Col440I_1, Col (BS512) 1, and ColpVC_1, indicates that this strain may be capable of producing colicins, bacteriocins that may suppress competing bacterial strains. These results demonstrate that this strain might be capable of horizontal gene transfer and the spread of antibiotic resistance by plasmids.
Table 3.
Plasmids predicted from whole genome sequencing (WGS) contigs of K. pneumoniae Kpn_R01 strain using PlasmidFinder (v2.1.6).
3.6. CRISPR-Cas Systems
The CRISPR-Cas Finder analysis revealed unique CRISPR arrays in two sequences, Seq31 and Seq37. Seq31 consisted of a single CRISPR array (Seq31_1) with a length of 139 bp, featuring 50 bp repeats and 40 bp spacers, demonstrating 98% conservation in both the repeats and spacers. Seq37 exhibited a significantly larger CRISPR array (Seq37_1) of 2649 bp, consisting of 29 bp repeats and 43 spacers, with a repeat conservation of 75.86% and a spacer conservation of 95.69%. Seq37 was oriented in the forward direction and exhibited an evidence level of 4, indicating a well-defined CRISPR locus. The findings demonstrate that although CRISPR elements are present in the dataset, their prevalence is low, with only a limited number of sequences showing functional or well-conserved CRISPR arrays (Table S2).
3.7. Prophage Regions and Viral Sequences
VirSorter analysis detected 14 potential prophage sequences in the sample, the majority of which were dsDNA phages, as well as one ssDNA phage (Seq129). The prophage sequences varied in length from 2300 bp (Seq129) to 49,050 bp (Seq34). The highest confidence values (maximum score = 1.00) were found in Seq34 and Seq43, indicating strong prophage signals. The number of signature genes ranged from 0 to 14, with Seq34 having the most. Multiple sequences, including Seq86 and Seq102, had low confidence scores (0.613 and 0.50, respectively) and lacked hallmark genes, indicating weaker prophage signatures. The discovery of these putative prophages provides information about potential viral components inside the investigated bacterial genomes, which may influence bacterial evolution and resistance mechanisms (Table S3).
3.8. Klebsiella Phage Kpn_R1 Isolation, Morphology, Assembly and Taxonomy
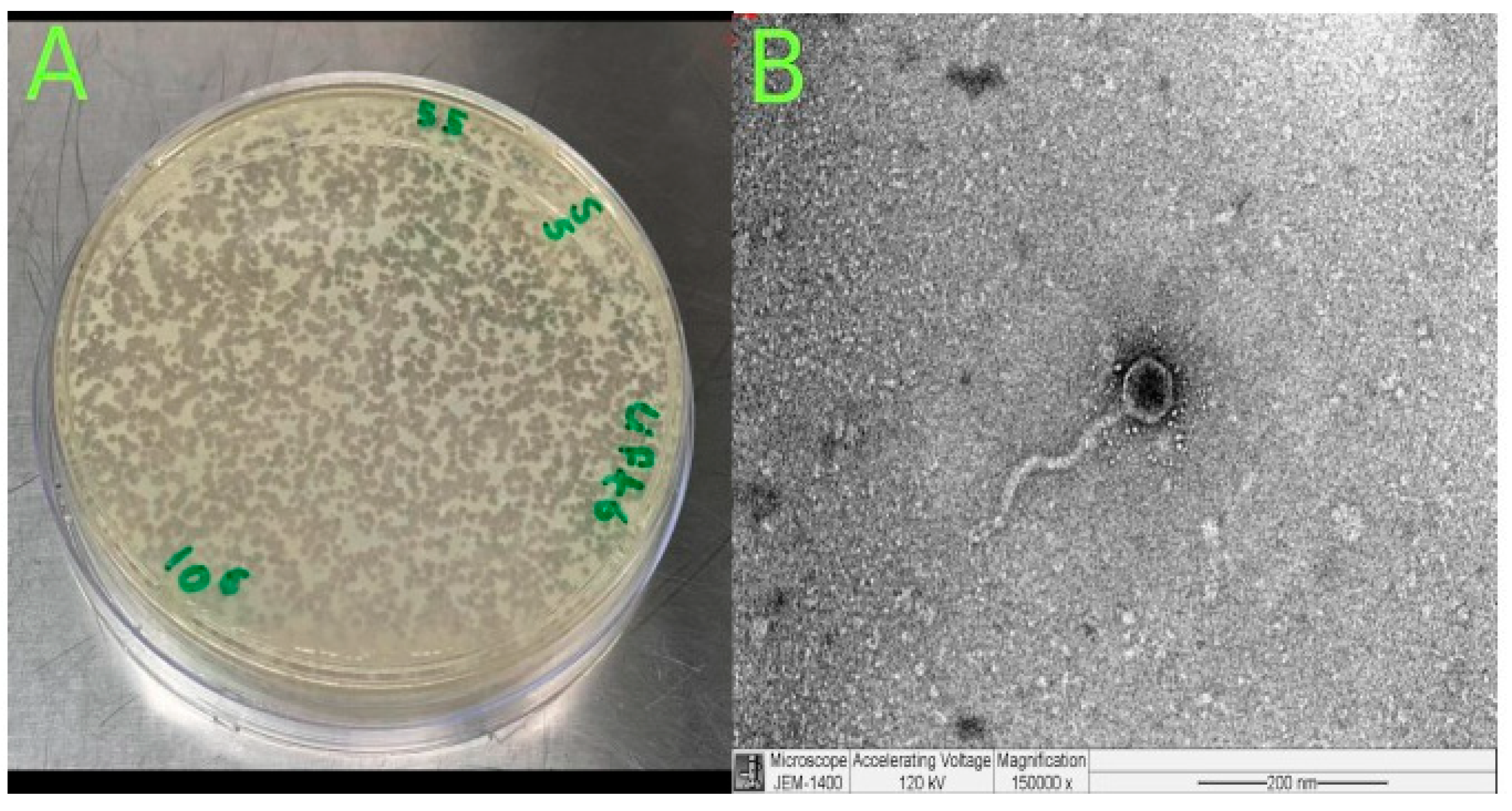
The phage was isolated from hospital wastewater with distinct plaque morphology (Figure 2A). TEM image examination showed that Klebsiella phage Kpn_R1 had a polyhedral head with a diameter of 53.7 nm and a wavy 178.3 nm long tail morphology (Figure 2B). This, pursuant to the International Committee on Taxonomy of Viruses taxonomy system, places them in the Demerecviridae family.
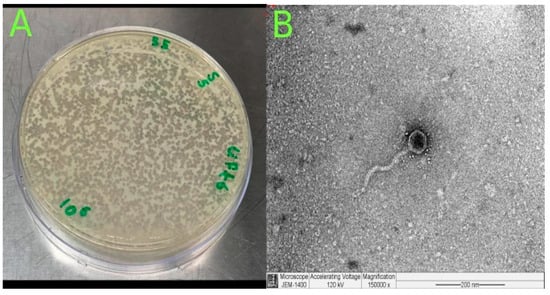
Figure 2.
Isolation and characterization of Klebsiella phage Kpn_R1. (A) Plaque morphology of Klebsiella phage Kpn_R1 on a bacterial lawn, showing distinct, clear plaques. (B) Transmission electron microscopy (TEM) image of Klebsiella phage Kpn_R1, revealing a polyhedral head (53.7 nm in diameter) and a wavy tail (178.3 nm in length). Based on morphology, the phage is classified within the Demerecviridae family according to the International Committee on Taxonomy of Viruses (ICTV). Scale bar: 200 nm.
3.9. General Genome Characteristics
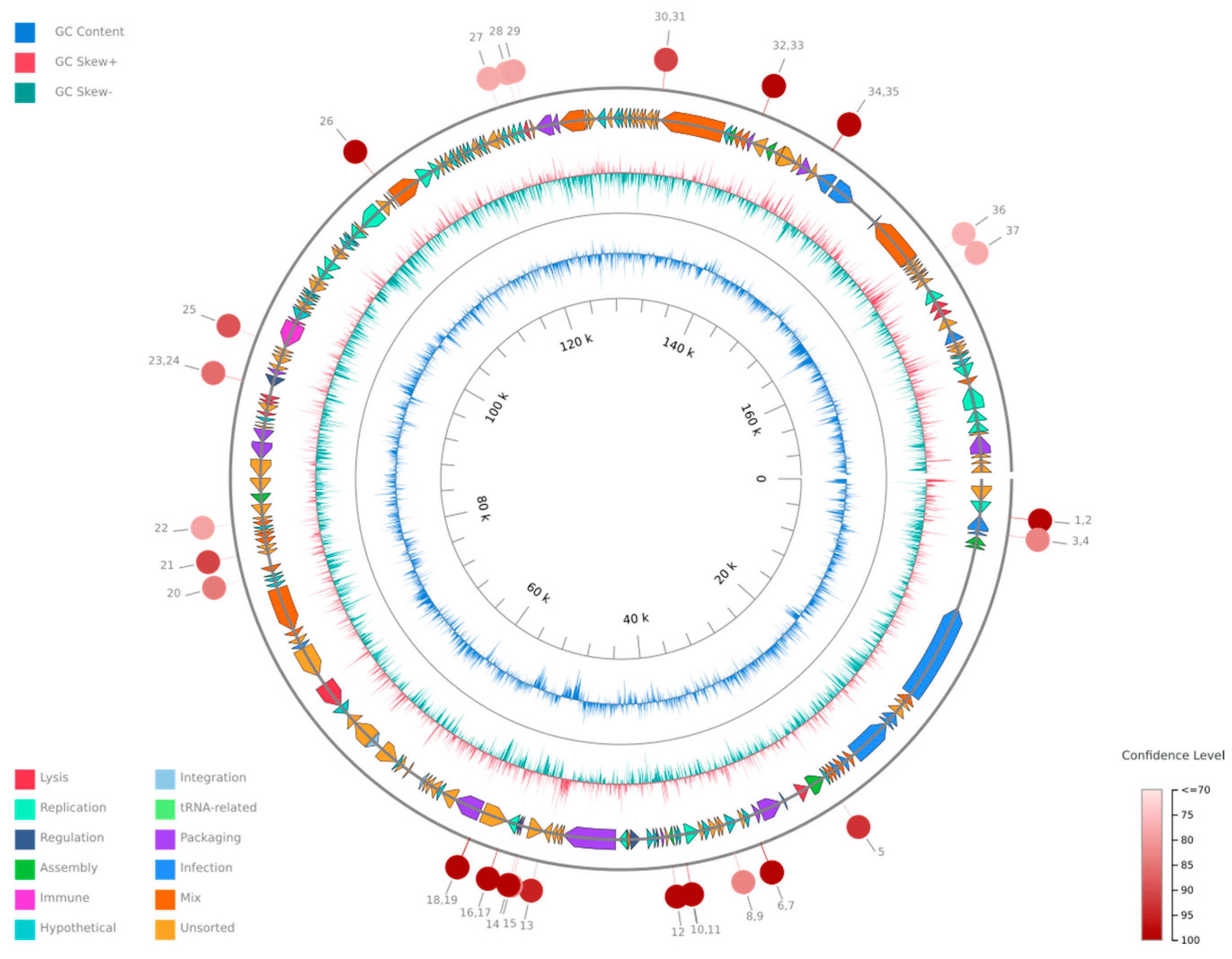
The phage genome is composed of 172,025 bp base pairs (bp), with a GC content of 46.3%. The genome consists of 211 predicted genes, with an average gene length of 639.47 bp. The genome type is classified as unknown, and it is of high quality with a 100.0% genome quality score with 100.0% completeness and 0.0% contamination.
The probability that the phage is temperate was estimated at 81%, based on the BACPHLIP database. No antibiotic resistance genes were identified within the genome, indicating that the phage does not haror any known antibiotic resistance traits, and the phage is confirmed not to be a provirus. The linearity of the phage genome was confirmed by PhageTerm. The genome consists of 220 predicted genes, with an average gene length of 639.47 bp. The phage genome organization is shown in Figure 3.
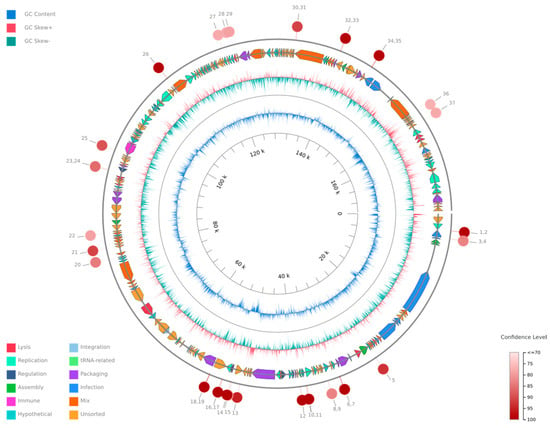
Figure 3.
A circular genome map of Klebsiella phage Kpn_R1 generated using PhageScope. The outermost rings represent annotated coding sequences (CDSs) classified by function, while the inner rings display GC content and GC skew. Red markers indicate putative virulence-associated or antibiotic resistance-related genes. The color gradient represents the variation in GC content across the genome.
3.10. Predicted Host Range, Evolutionary Relationships, and Genomic Similarity of Klebsiella Phage Kpn_R1
The predicted hosts of the annotated phage were inferred based on the best-matching phage genomes and their reported hosts. As shown in Table 4, the majority of hits (33 hits) were associated with Klebsiella, particularly K. pneumoniae, with an average genetic distance of 0.06, an average matching hash score of 215.3, and an average p-value of hits 3.38 × 10−111. This strong association suggests that K. pneumoniae is the most likely host for the annotated phage. Other potential hosts include Yersinia (2 hits), Salmonella (13 hits), Erwinia (1 hit), and Campylobacter (1 hit), though these associations are weaker, as indicated by higher genetic distances (0.22–0.23).
Table 4.
Predicted hosts as inferred through the best-matching phage genomes and their reported hosts.
Table 5 shows that the analysis grouped the closest relatives into several genera, with the highest number of hits belonging to Sugarlandvirus (20 hits) and Epseptimavirus (15 hits). The Sugarlandvirus genus showed the lowest average genetic distance (0.04) and the highest average matching hashes (250.8), indicating a strong genomic similarity. These results suggest that the isolated phage belongs to Caudoviricetes; Demerecviridae; Sugarlandvirus genus.
Table 5.
Taxonomic classification and genetic similarity metrics of Klebsiella phage Kpn_R1 and its closest relative genera (using the MASH algorithm), All hits with genetic distance < 1 against published phage genomes grouped by genus.
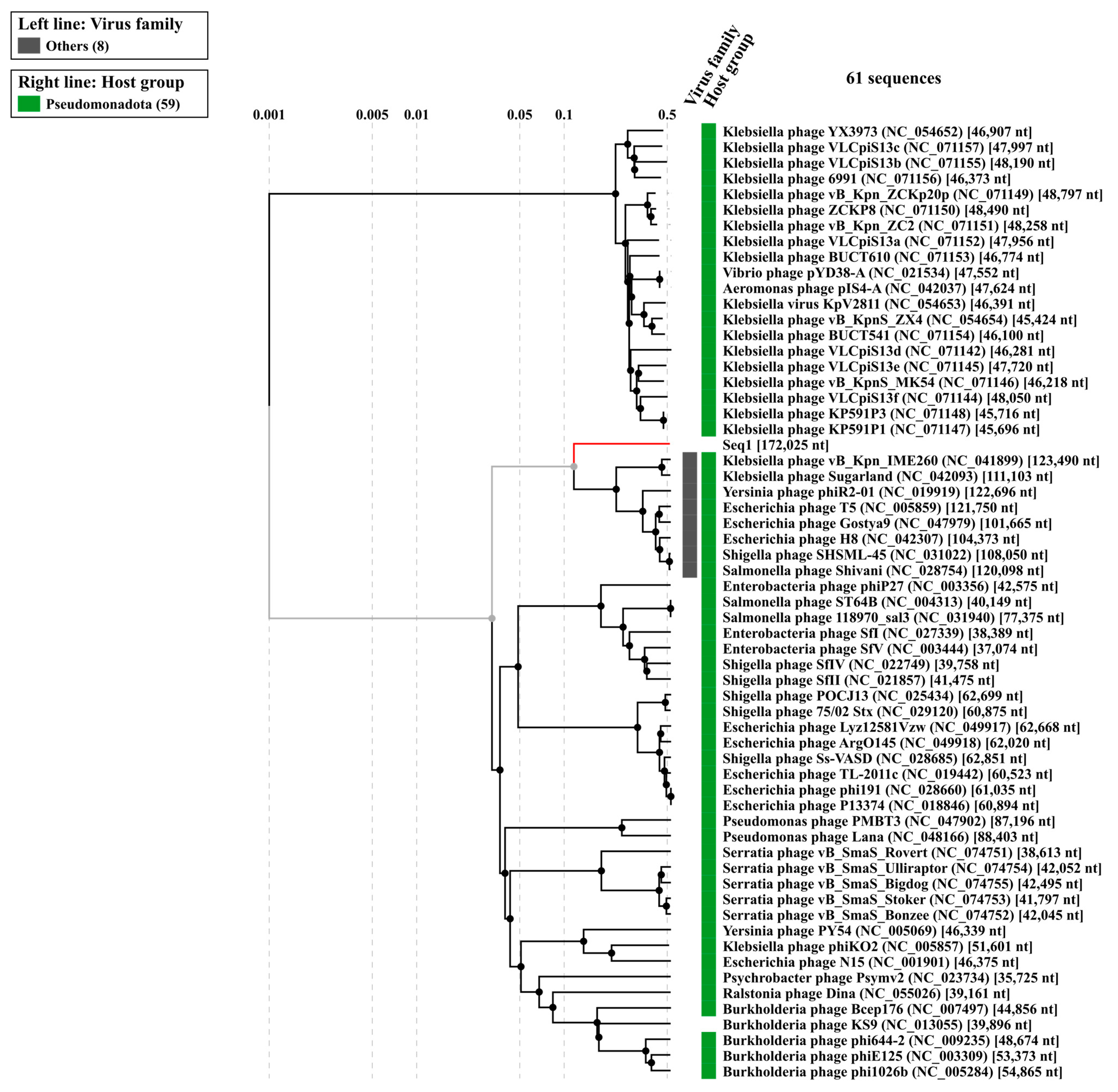
The evolutionary relationships of Klebsiella phage Kpn_R1 were investigated using whole-genome sequencing comparisons (Figure 4). The circular phylogenetic tree shows different clusters of closely related phages, with Kpn_R1 located next to Klebsiella phage vB_Kpn_IME260 and Klebsiella phage Sugarland. Kpn_R1 shares 92.8% genomic identity across 31.5% of its sequence with vB_Kpn_IME260, as well as 92.3% identity over 30.1% of its genome with Klebsiella phage Sugarland. The study revealed Escherichia phage N15, which had a mean identity of 69.8% across 14% of the genome, indicating a significant evolutionary difference.
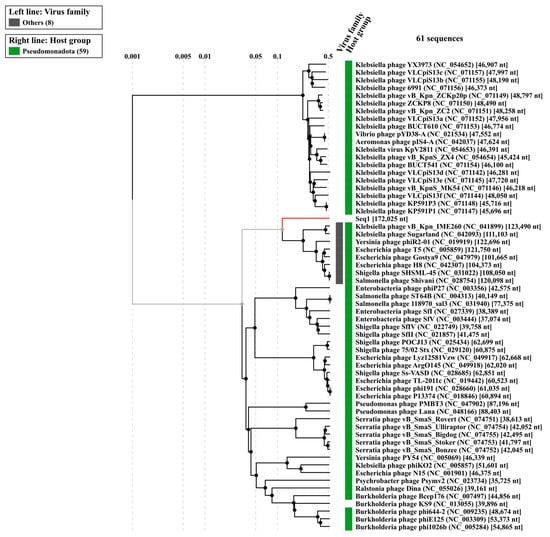
Figure 4.
The phylogenetic analysis of Klebsiella phage Kpn_R1 was conducted using ViPTree, a web server that generates viral proteomic trees based on genome-wide sequence similarities computed by tBLASTx. The circular proteomic tree illustrates the evolutionary relationships of Kpn_R1, highlighting its closest related phages forming distinct clusters (gray).
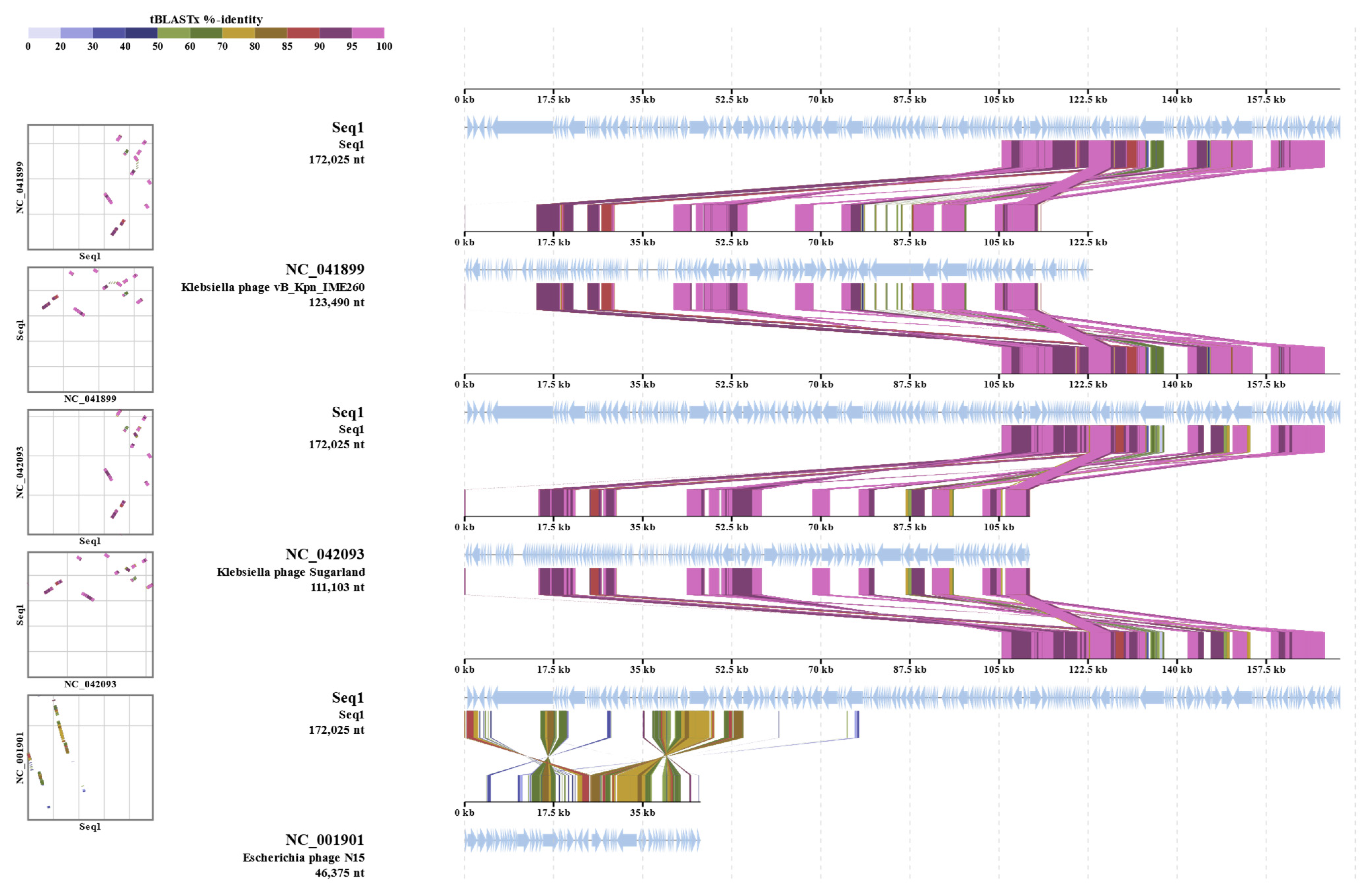
The alignment of protein sequences (Figure 5) further highlights the similarities between Kpn_R1 and its closest relatives. The strongest sequence similarities between Klebsiella phage Kpn_R1 (Seq1), Klebsiella phage vB_Kpn_IME260, and Klebsiella phage Sugarland are depicted by connected syntenic blocks, with the most conserved regions shown in pink. The distinct structural organization of Escherichia phage N15 further reinforces its evolutionary divergence.
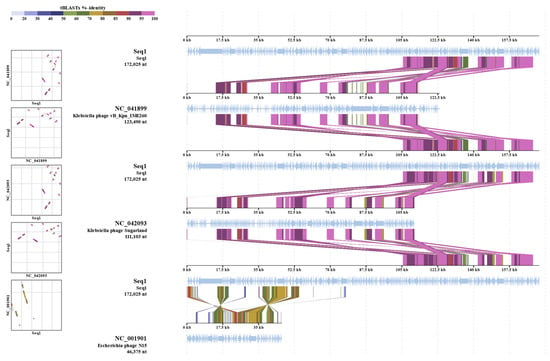
Figure 5.
Proteomic similarity map of Klebsiella phage Kpn_R1 and related phages. The alignment shows protein sequence similarities between Klebsiella phage Kpn_R1 (Seq1) and its closest relatives: Klebsiella phage vB_Kpn_IME260, Klebsiella phage Sugarland, and Escherichia phage N15. Conserved regions are depicted by connected syntenic blocks, with the strongest similarities shown in pink. The distinct structural organization of Escherichia phage N15 highlights its evolutionary divergence.
As shown in the Table S4, the whole-genome sequencing of the isolated phage demonstrated a 92.8% identity across 31.5% of the whole genome of its closest similar Klebsiella phage vB_Kpn_IME260. Klebsiella phage Sugarland (NC_042093) exhibited a mean identity of 92.3% across 30.1% of the genome length, indicating a modest degree of similarity with related phages. Sugarland is classified within the Pseudomonadota category, signifying a wider host range among Enterobacteriaceae members.
The comparison also recognized Escherichia phage N15 (NC_001901) as a similar phage; however, it had a lower mean identity of 69.8% across merely 14% of the genome. Different from lytic phages such as Sugarland, N15 is a completely defined temperate phage that may integrate into the bacterial genome as a linear plasmid. The somewhat low identity and genome coverage suggest that N15 is evolutionarily different even if it shares certain genetic elements with the isolated phage. These findings suggest that the isolated phage may adapt its genome for better survival or bacterial genome evolution via horizontal gene transfer technologies due to its evolutionary links and functional capacity. Comparative genomics will dominate future research to identify distinct genetic variables influencing host specificity and lytic activity.
3.11. Klebsiella Phage Kpn_R1 Genome Annotation
The 220 ORFs (Table S4) in the annotated phage genome have been categorized into functional categories based on their anticipated functions. As shown in Table 6, hypothetical/uncharacterized proteins comprise the majority of the total ORFs, accounting for 48.34% (102 ORFs). These proteins are not known to have any functions or exhibit any significant similarities to known proteins. This implies that there are novel genes that are unique to phages and may play distinct functions in the phage lifecycle. Structural proteins constitute the second-largest group, which accounts for 18.01% (38) of the ORFs. This encompasses the primary capsid protein, tail fiber proteins, and portal proteins, all of which are crucial for the formation of virion structures. The significant presence of structural proteins highlights an accurate virion assembly for effective infection and replication. Proteins associated with DNA replication and metabolism comprise 11.85% (25) of the open reading frames (ORFs). DNA polymerase, helicase, exonuclease, and recombination mediators are all in this group. These components are essential for the preservation and replication of genomes. The phage’s ability to modify host DNA and facilitate effective genome replication is emphasized by the presence of these proteins. Transcriptional regulators, anti-termination proteins, and XRE family regulators comprise 2.84% (6) of the ORFs. It is probable that these proteins are involved in the regulation of gene expression throughout the phage lifecycle, which allows for the rapid activation or suppression of genes that are necessary for the various stages of infection.
Table 6.
Functional categorization of Klebsiella Phage Kpn_R1 open reading frames (ORFs).
The host lysis group, which comprises endolysin, holin, and spanin proteins, comprises 1.42% (3) of the ORFs. These proteins are crucial for lysing the host cell to liberate freshly formed phage particles, signifying the concluding phase of the lytic cycle. The category of Moron, auxiliary metabolic genes, and host takeover comprise 6.16% (13) of the ORFs. Also included in the genome annotated ORFs are the ParA/B-like proteins comprising 1.42% (3). These proteins participate in plasmid partitioning with protelomerase presence, indicating a mechanism for the steady inheritance of the phage genome throughout cellular division, akin to the N15 phage and protelomerase reinforces this similarity since protelomerase is characteristic of N15’s replication and maintenance approach, facilitating telomere resolution and linear genome preservation. The genome encodes a toxin–antitoxin (TA) system, especially a RelE/ParE family toxin (Table S3). Plasmid maintenance, stress response, and persistence mechanisms are frequently implemented by TA systems. In phages, they may influence the behavior of host cells to promote phage survival or facilitate the stability of the prophage state. The phage genome exhibits a functional distribution that is typical of temperate phages, with a focus on host lysis, DNA replication, and structural assembly. The extensive unexplored biology is suggested by the significant predominance of putative proteins. However, the presence of partitioning proteins, protelomerase, and a toxin–antitoxin system suggests a reproductive and maintenance strategy similar to that of the N15 phage. In order to confirm the mechanisms of the phage’s lifecycle and to elucidate the functions of the potential proteins, additional experimental investigations are required.
4. Discussion
In the current study, K. pneumoniae Kpn-R1 strain was isolated from an ICU patient’s sputum sample. The strain’s genome size was 5,835,643 bp, with a GC content of 56.82%, according to whole genome sequencing. The strain was identified as a high-risk ST 147 clone using Pasteur Institute MLST. The genome size of K. pneumoniae ST147 isolates is generally between 5.0 and 5.5 Mbp and a GC content range of 56–58% [2]. These genomic traits reflect the species’ flexibility and ability to accept foreign genetic material, such as plasmids and integrons, which typically carry antibiotic resistance genes [90].
Kpn_R01 strain exhibits an XDR profile influenced by 32 AMR spanning multiple antibiotic classes, including beta-lactams, fluoroquinolones, aminoglycosides, tetracyclines, macrolides, phenolics, polymyxins, and sulfonamides. According to Nordmann et al. (2011) and Logan and Weinstein (2017), there is a wide resistance pattern associated with the increasing incidence of hospital-acquired K. pneumoniae bacteria that are resistant to several drugs [4,91]. The research revealed that the Kpn_R01 genome contains beta-lactamase resistance genes including blaSHV-11, blaNDM-1, OmpK37, and MdtQ. SHV-11 beta-lactamase and NDM-1 carbapenemase are the primary enzymes for penicillin, cephalosporin, and carbapenem degradation [92]. Alterations in porins (OmpK37) and efflux (MdtQ) impede the entry of beta-lactamase into the bacterial cell by reducing cell permeability [93]. ST147 is a high-risk clone that has been extensively documented and is associated with carbapenem resistance, notably as a result of the production of carbapenemases like NDM-1, SHV-11, and OXA-48 [94]. Studies undertaken in Saudi Arabia, UAE, and Oman have highlighted the frequency of ST147 in clinical settings, with a significant percentage of isolates carrying the blaNDM-1 and blaOXA-48 genes [95,96,97].
Resistance to fluoroquinolones, including ciprofloxacin (≥4 μg/mL) and levofloxacin (≥8 μg/mL), is facilitated by a synergy of efflux pumps (oqxA, acrA, marA). These methods diminish intracellular drug concentrations and modify drug-binding sites, hence imparting significant resistance to this family of antibiotics [98]. The strain’s resistance to aminoglycosides, such as gentamicin (≥16 μg/mL) and amikacin (≥64 μg/mL), is mediated by 16S rRNA methyltransferase (armA) and aminoglycoside-modifying enzymes (aadA2) [99]. Efflux pumps (tet(A), tet(B)), and ribosomal protection proteins (tet(M)) mediate resistance to tetracyclines (tetracycline ≥16 μg/mL, tigecycline ≥2 μg/mL), which reduce intracellular drug concentrations or protect the ribosome from inhibition [89,100]. Similarly, efflux transporters (msrE, mphE) are mechanisms that have been increasingly observed in Gram-negative bacteria [101] that facilitate resistance to macrolides (erythromycin ≥8 μg/mL, azithromycin ≥16 μg/mL). The strain’s resistance to trimethoprim (≥16 μg/mL) and sulfamethoxazole (≥256 μg/mL) is mediated by dihydropteroate synthase (sul1) and dihydrofolate reductase (dfrA12), respectively. The drug targets are altered by these enzymes, which prevent the inhibition of folate biosynthesis, a critical pathway for bacterial proliferation, thereby conferring resistance [102]. Similarly, fosfomycin thiol transferase (fosA5) is responsible for the inactivation of the antibiotic, resulting in resistance to fosfomycin (≥64 μg/mL) [103]. Phosphoethanolamine transferase (arnT, eptB) and mutations in the mgrB gene are associated with resistance to colistin and polymyxin B (≥4 μg/mL). These mutations modify the lipid A component of lipopolysaccharide, thereby reducing the binding affinity of polymyxins [104]. This mechanism is particularly alarming as polymyxins are frequently employed as last-resort antibiotics to treat XDR Gram-negative infections. The efflux transporters (qacEdelta1, leuO) that are frequently associated with biocide resistance in Gram-negative bacteria facilitate the strain’s resistance to quaternary ammonium compounds (≥50 μg/mL) [105]. The strain’s persistence in hospital environments, where biocides are frequently employed for disinfection, may be influenced by this resistance mechanism [74].
In addition to its extensive resistance profile, the strain possessed several virulence factors that exacerbated its pathogenesis. The E. coli common pilus (ECP) gene cluster implicates the strain’s capacity to adhere to host tissues and form biofilms, which is a critical factor in the establishment of infections [106]. Enterobactin genes and the yersiniabactin biosynthetic gene cluster suggest that the strain is capable of acquiring iron, a critical nutrient for bacterial survival and virulence in the host [107]. Outer membrane protein A has been demonstrated to safeguard microorganisms from host immune responses [108], which further supports the notion that the ompA gene has immune evasion capabilities. These virulence factors collectively contribute to the strain’s capacity to cause severe infections, particularly in immunocompromised patients.
The strain has a non-operational Type I-E CRISPR-Cas gene cluster, diminishing its tolerance to foreign genetic elements, such as plasmids and phages, hence facilitating horizontal gene transfer (HGT) and the dissemination of antibiotic resistance genes [109]. This property is characterized by its plasmid content, including In-cHI1B_1_pNDM-MAR, IncFIB (K)_1_Kpn3, and IncFIB (pKPHS1)_1_pKPHS1 replicons [110]. The strain’s ability to generate bacteriocins is shown by the presence of colicin genes (Col440I_1, Col (BS512)_1, ColpVC_1), potentially augmenting its survival in polymicrobial settings and suppressing the proliferation of rival bacterial strains [111]. Eleven prophage regions were also detected, which often harbor genes that enhance bacterial virulence and antibiotic resistance, highlighting the genetic diversity of strains and their potential for horizontal gene transfer [5]. Eleven prophage regions were also detected, which often harbor genes that enhance bacterial virulence and antibiotic resistance, highlighting the genetic diversity of the strain and its potential for horizontal gene transfer [8]. This property makes this Middle Eastern ST147 a uniquely adaptable nosocomial pathogen.
Of particular interest is the isolation of an N15-like temperate phage from hospital wastewater using the Kpn-R1 strain as a host. This study’s significant development is the characterization of a N15-like temperate phage exhibiting a linear plasmid prophage lifestyle [9], a replication method seldom reported in K. pneumoniae phages. This phage, in contrast to other phages often linked to ST147, utilizes protelomerase (93.3% similarity to N15 TelN) and ParA/B partitioning proteins to sustain lysogeny, resembling the N15 phage’s capacity to exist as a linear plasmid [9,11,112]. The incorporation of a RelE/ParE toxin–antitoxin (TA) system within the phage genome further differentiates it from previous accounts of N15-like pKO2 linear plasmid prophage of K. oxytoca [14]. The presence of TA systems, usually associated with plasmid stability, indicates a unique function in phage-mediated stress adaptation and prophage survival [113,114]. These results enhance the understanding of phage roles in bacterial evolution, especially in high-risk clones such as ST147 where prophages may serve as repositories for resistance or virulence genes.
The structural proteins identified in the genome, including major capsid proteins, tail fibers, and portal proteins, are essential for virion assembly and host recognition. These proteins are highly conserved among phages and play a critical role in the infection cycle [115,116]. The regulatory proteins identified in the genome, including transcriptional regulators and anti-termination proteins, ensure the timely activation or repression of genes required for different stages of infection, such as lysogeny and lytic replication [117]. The UmuD-like protein identified in the genome is part of the SOS response system, which is involved in DNA damage repair and mutagenesis [118,119]. This finding is consistent with studies on other phages where UmuD homologs have been shown to enhance survival in hostile environments [88].
This study uniquely associates CRISPR inactivation with MGE-driven evolution in Middle Eastern clones, a feature previously linked to plasmid loss in other lineages [89]. Furthermore, we provide the first evidence of an N15-like phage containing TA systems in ST147, contesting the notion that phage-mediated adaptation in K. pneumoniae is restricted to lytic or temperate phages. These results reclassify ST147 as a paradigm for investigating region-specific pathogen evolution and phage–host coadaptation.
This study bridges a critical gap by characterizing the first N15-like phage within the high-risk ST147 clone, revealing its unique TA system and replication strategy. By linking clinical and environmental reservoirs, we propose actionable strategies for phage engineering and surveillance to combat XDR K. pneumoniae in healthcare settings. These findings shift the paradigm from observational genomics to translational solutions targeting phage–bacterial coevolution.
5. Conclusions
This study presents the genomic interaction between a novel N15-like temperate phage sourced from hospital wastewater and an extremely drug-resistant K. pneumoniae ST147 strain isolated from an ICU patient, thereby revealing significant new insights into resistance dissemination and phage-mediated evolution. The ST147 strain’s hybrid plasmid (IncHI1B_1_pNDM-MAR/IncFIB), which carries blaNDM-1 and colicin genes, along with CRISPR-Cas inactivation and 11 prophage regions, highlights its genetic plasticity and potential nosocomial threat. The linear plasmid-like replication system of the phage, along with protelomerase and a novel RelE/ParE toxin–antitoxin (TA) system—previously unreported in temperate Klebsiella phages—highlights mechanisms that enhance resistance traits and promote phage survival.
Tracking phage-mediated resistance gene transfer between clinical and environmental reservoirs helps us show how wastewater monitoring may prevent outbreaks. By suggesting actionable plans, this work goes beyond the current genomic cataloging of ST147: the phage’s replication machinery provides a template for engineered therapies (e.g., CRISPR-Cas delivery), while the strain’s regional resistance profile calls for tailored antibiotic stewardship to reduce last-resort drug misuse. Emphasizing phage–bacterial interactions as a frontier in the fight against XDR infections, our results change the paradigm from passive genomic monitoring to proactive, ecology-driven therapies. Future research should confirm the functional significance of the TA system and investigate phage-based diagnostics to interfere with resistance transfer in healthcare environments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms13040908/s1. Table S1: The key findings from the assembly analysis of O-antigen and capsular type; Table S2: CRISPR-Cas Finder Results for Identified Sequences; Table S3: Prophages Identified Within the Genome of Klebsiella pneumoniae Kpn_R01; Table S4: Functional annotation of Klebsiella phage Kpn_R1 genome; Table S5: Genome similarity analysis of Klebsiella phage Kpn_R1 with other bacteriophages. The table presents comparative genomic data, including sequence identity, alignment length, and taxonomic classification of similar phages.
Author Contributions
Conceptualization, R.Y.; methodology, R.Y., A.A. (Aljawharah Albaqami), A.A. (Amal ALzahrani), M.A., E.T.A. and M.A.A.-M.; software, R.Y.; validation, R.Y.; formal analysis, N.A. (Nada Alhazmi); investigation, R.Y. and M.A.A.-M.; resources, N.A. (Najwa Alharbi); bioinformatics analysis and writing—original draft, S.M.A.; writing—review and editing, R.Y.; supervision, R.Y.; project administration, R.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by King Abdullah International Medical Research Center (KAIMRC) in Riyadh, Saudi Arabia grant number NRC21R/053/02.
Institutional Review Board Statement
This study was conducted in conformity with ethical policies concerning the use of microbial genetic data. The examined K. pneumoniae strain came from a clinical sample taken with informed permission and approved by the relevant institutional review board (IRB) NRC21R/053/02 (24 February 2021). Patients’ right to privacy and confidentiality was fiercely maintained to guarantee that the research included no personally identifying data whatsoever. Complying with relevant national and international ethical research norms, the study adhered to the criteria established in the Declaration of Helsinki.
Informed Consent Statement
Not Applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, A.A.; Alwashmi, A.S.S.; Abalkhail, A.; Alkahtani, A.M. Emerging Challenges in Klebsiella pneumoniae: Antimicrobial Resistance and Novel Approach. Microb. Pathog. 2025, 202, 107399. [Google Scholar] [CrossRef] [PubMed]
- Wyres, K.L.; Lam, M.M.C.; Holt, K.E. Population Genomics of Klebsiella pneumoniae. Nat. Rev. Microbiol. 2020, 18, 344–359. [Google Scholar] [CrossRef]
- Effah, C.Y.; Sun, T.; Liu, S.; Wu, Y. Klebsiella pneumoniae: An Increasing Threat to Public Health. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Naas, T.; Poirel, L. Global Spread of Carbapenemase-Producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Riwu, K.H.P.; Effendi, M.H.; Rantam, F.A.; Khairullah, A.R.; Widodo, A. A Review: Virulence Factors of Klebsiella pneumonia as Emerging Infection on the Food Chain. Vet. World 2022, 15, 2172–2179. [Google Scholar] [CrossRef]
- Russo, T.A.; Marr, C.M. Hypervirulent Klebsiella pneumoniae. Clin. Microbiol. Rev. 2019, 32, e00001-19. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal Gene Transfer and Adaptive Evolution in Bacteria. Nat. Rev. Microbiol. 2022, 20, 206–218. [Google Scholar] [CrossRef]
- Fortier, L.-C.; Sekulovic, O. Importance of Prophages to Evolution and Virulence of Bacterial Pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef]
- Ravin, V.; Ravin, N.; Casjens, S.; Ford, M.E.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequence and Analysis of the Atypical Temperate Bacteriophage N15. J. Mol. Biol. 2000, 299, 53–73. [Google Scholar] [CrossRef]
- Ravin, N.V. N15: The Linear Phage–Plasmid. Plasmid 2011, 65, 102–109. [Google Scholar] [CrossRef]
- Ravin, N.V. Mechanisms of Replication and Telomere Resolution of the Linear Plasmid Prophage N15. FEMS Microbiol. Lett. 2003, 221, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ravin, N.V. Replication and Maintenance of Linear Phage-Plasmid N15. Microbiol. Spectr. 2015, 3, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B.; Huang, W.M.; Bunny, K.L.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The pKO2 Linear Plasmid Prophage of Klebsiella oxytoca. J. Bacteriol. 2004, 186, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M.J. Engineering Therapeutic Phages for Enhanced Antibacterial Efficacy. Curr. Opin. Virol. 2022, 52, 182–191. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage Host Range and Bacterial Resistance. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 70, pp. 217–248. ISBN 978-0-12-380991-9. [Google Scholar]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-Specific Antimicrobials Using Efficiently Delivered RNA-Guided Nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4629–4634. [Google Scholar] [CrossRef]
- Mahler, M.; Costa, A.R.; Van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for Bacteriophage Genome Engineering. Trends Biotechnol. 2023, 41, 669–685. [Google Scholar] [CrossRef]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef]
- Aprea, G.; D’Angelo, A.R.; Prencipe, V.A.; Migliorati, G. Bacteriophage Morphological Characterization by Using Transmission Electron Microscopy. JLS 2015, 10, 214–220. [Google Scholar] [CrossRef]
- Moller, A.G.; Lindsay, J.A.; Read, T.D. Determinants of Phage Host Range in Staphylococcus Species. Appl. Environ. Microbiol. 2019, 85, e00209-19. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Interpolated Variable Order Motifs for Identification of Horizontally Acquired DNA: Revisiting the Salmonella Pathogenicity Islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, gkz935. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic Data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Starikova, E.V.; Tikhonova, P.O.; Prianichnikov, N.A.; Rands, C.M.; Zdobnov, E.M.; Ilina, E.N.; Govorun, V.M. Phigaro: High-throughput prophage sequence annotation. Bioinformatics 2020, 36, 3882–3884. [Google Scholar] [CrossRef]
- Brown, C.L.; Mullet, J.; Hindi, F.; Stoll, J.E.; Gupta, S.; Choi, M.; Keenum, I.; Vikesland, P.; Pruden, A.; Zhang, L. mobileOG-Db: A manually curated database of protein families mediating the life cycle of bacterial mobile genetic elements. Appl. Environ. Microbiol. 2021, 88, e00991-22. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Wick, R.R.; Heinz, E.; Holt, K.E.; Wyres, K.L. Kaptive Web: User-Friendly Capsule and Lipopolysaccharide Serotype Prediction for Klebsiella Genomes. J. Clin. Microbiol. 2018, 56, e00197-18. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.Org Website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap: Rapid Ribosomal RNA Prediction [Internet]. 2013. Available online: https://github.com/tseemann/barrnap (accessed on 9 March 2025).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A Tool for Fast and Accurate Determination of Phage Termini and Packaging Mechanism Using next-Generation Sequencing Data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef] [PubMed]
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a fast and Accurate tool and web server to classify phage structural proteins. PLoS Comput. Biol. 2020, 16, e1007845. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of large-scale biases in the current collection of cultured phage genomes. Phage 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using The Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, D.; Xu, G.; Huang, W.; Wang, X. Molecular epidemiology and drug resistant mechanism in carbapenem-resistant Klebsiella pneumoniae isolated from pediatric patients in Shanghai, China. PLoS ONE 2018, 13, e0194000. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Dortet, L.; Bernabeu, S.; Nordmann, P. Genetic Features of blaNDM-1 -Positive Enterobacteriaceae. Antimicrob. Agents Chemother. 2011, 55, 5403–5407. [Google Scholar] [CrossRef]
- Bulman, Z.P.; Krapp, F.; Pincus, N.B.; Wenzler, E.; Murphy, K.R.; Qi, C.; Ozer, E.A.; Hauser, A.R. Genomic Features Associated with the Degree of Phenotypic Resistance to Carbapenems in Carbapenem-Resistant Klebsiella pneumoniae. mSystems 2021, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, X.; Ye, L.; Hou, T.; Liu, Y.; Liu, G.; Wang, Q.; Zhang, D. Hypermucoviscous Multidrug-Resistant Klebsiella variicola Strain LL2208 Isolated from Chinese Longsnout Catfish (Leiocassis longirostris): Highly Similar to Human K. variicola Strains. Pathogens 2024, 13, 647. [Google Scholar] [CrossRef]
- Schneiders, T.; Amyes, S.G.B.; Levy, S.B. Role of AcrR and RamA in Fluoroquinolone Resistance in Clinical Klebsiella pneumoniae Isolates from Singapore. Antimicrob. Agents Chemother. 2003, 47, 2831–2837. [Google Scholar] [CrossRef]
- Yamasaki, E.; Yamada, C.; Jin, X.; Nair, G.B.; Kurazono, H.; Yamamoto, S. Expression of marA is remarkably increased from the early stage of development of fluoroquinolone-resistance in uropathogenic Escherichia coli. J. Infect. Chemother. 2015, 21, 105–109. [Google Scholar] [CrossRef]
- Moosavian, M.; Khoshkholgh Sima, M.; Ahmad Khosravi, N.; Abbasi Montazeri, E. Detection of OqxAB Efflux Pumps, a Multidrug-Resistant Agent in Bacterial Infection in Patients Referring to Teaching Hospitals in Ahvaz, Southwest of Iran. Int. J. Microbiol. 2021, 2021, 2145176. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Steinberg, D. Targeting the Holy Triangle of Quorum Sensing, Biofilm Formation, and Antibiotic Resistance in Pathogenic Bacteria. Microorganisms 2022, 10, 1239. [Google Scholar] [CrossRef]
- Zhou, Y.; Ai, W.; Guo, Y.; Wu, X.; Wang, B.; Xu, Y.; Rao, L.; Zhao, H.; Wang, X.; Yu, F. Co-Occurrence of Rare ArmA-, RmtB-, and KPC-2–Encoding Multidrug-Resistant Plasmids and Hypervirulence iuc Operon in ST11-KL47 Klebsiella pneumoniae. Microbiol. Spectr. 2022, 10, e02371-21. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Cave, R.; Ter-Stepanyan, M.M.; Kotsinyan, N.; Chen, J.; Zhang, L.; Jiang, T.; Mkrtchyan, H.V. Whole-Genome Sequencing and Comparative Genomics Analysis of a Newly Emerged Multidrug-Resistant Klebsiella pneumoniae Isolate of ST967. Microbiol. Spectr. 2023, 11, e04011-22. [Google Scholar] [CrossRef]
- Gravey, F.; Sévin, C.; Castagnet, S.; Foucher, N.; Maillard, K.; Tapprest, J.; Léon, A.; Langlois, B.; Le Hello, S.; Petry, S. Antimicrobial resistance and genetic diversity of Klebsiella pneumoniae strains from different clinical sources in horses. Front. Microbiol. 2024, 14, 1334555. [Google Scholar] [CrossRef]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and Outlook on Enzyme-Mediated Macrolide Resistance. Front. Microbiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, A.C.; Stogios, P.J.; Koteva, K.; Skarina, T.; Evdokimova, E.; Savchenko, A.; Wright, G.D. The evolution of substrate discrimination in macrolide antibiotic resistance enzymes. Nat. Commun. 2018, 9, 112. [Google Scholar] [CrossRef]
- Talat, A.; Khan, F.; Khan, A.U. Genome analyses of colistin-resistant high-risk blaNDM-5 producing Klebsiella pneumoniae ST147 and Pseudomonas aeruginosa ST235 and ST357 in clinical settings. BMC Microbiol. 2024, 24, 174. [Google Scholar] [CrossRef]
- Venkatesan, M.; Fruci, M.; Verellen, L.A.; Skarina, T.; Mesa, N.; Flick, R.; Pham, C.; Mahadevan, R.; Stogios, P.J.; Savchenko, A. Molecular mechanism of plasmid-borne resistance to sulfonamide antibiotics. Nat. Commun. 2023, 14, 4031. [Google Scholar] [CrossRef]
- Wang, W.; Baloch, Z.; Peng, Z.; Hu, Y.; Xu, J.; Fanning, S.; Li, F. Genomic characterization of a large plasmid containing a bla NDM-1 gene carried on Salmonella enterica serovar Indiana C629 isolate from China. BMC Infect. Dis. 2017, 17, 479. [Google Scholar] [CrossRef]
- Tomich, A.D.; Klontz, E.H.; Deredge, D.; Barnard, J.P.; McElheny, C.L.; Eshbach, M.L.; Weisz, O.A.; Wintrode, P.; Doi, Y.; Sundberg, E.J.; et al. Small-Molecule Inhibitor of FosA Expands Fosfomycin Activity to Multidrug-Resistant Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2019, 63, e01524-18. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gong, L.; Liu, E.; Li, C.; Wang, Y.; Liang, J. Characterization of the Disinfectant Resistance Genes qacEΔ1 and cepA in Carbapenem-Resistant Klebsiella pneumoniae Isolates. Am. J. Trop. Med. Hyg. 2024, 110, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Linde, J.; Hammer, P.; Pletz, M.W.; Neubauer, H.; Sprague, L.D. WGS-Based Phenotyping and Molecular Characterization of the Resistome, Virulome and Plasmid Replicons in Klebsiella pneumoniae Isolates from Powdered Milk Produced in Germany. Microorganisms 2022, 10, 564. [Google Scholar] [CrossRef]
- Shankar, C.; Vasudevan, K.; Jacob, J.J.; Baker, S.; Isaac, B.J.; Neeravi, A.R.; Sethuvel, D.P.M.; George, B.; Veeraraghavan, B. Hybrid Plasmids Encoding Antimicrobial Resistance and Virulence Traits Among Hypervirulent Klebsiella pneumoniae ST2096 in India. Front. Cell. Infect. Microbiol. 2022, 12, 875116. [Google Scholar] [CrossRef]
- Shukla, S.; Desai, S.; Bagchi, A.; Singh, P.; Joshi, M.; Joshi, C.; Patankar, J.; Maheshwari, G.; Rajni, E.; Shah, M.; et al. Diversity and Distribution of β-Lactamase Genes Circulating in Indian Isolates of Multidrug-Resistant Klebsiella pneumoniae. Antibiotics 2023, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Stercz, B.; Farkas, F.B.; Tóth, Á.; Gajdács, M.; Domokos, J.; Horváth, V.; Ostorházi, E.; Makra, N.; Kocsis, B.; Juhász, J.; et al. The influence of antibiotics on transitory resistome during gut colonization with CTX-M-15 and OXA-162 producing Klebsiella pneumoniae ST15. Sci. Rep. 2021, 11, 6335. [Google Scholar] [CrossRef]
- Salloum, T.; Arabaghian, H.; Alousi, S.; Abboud, E.; Tokajian, S. Genome sequencing and comparative analysis of an NDM-1-producing Klebsiella pneumoniae ST15 isolated from a refugee patient. Pathog. Glob. Health 2017, 111, 166–175. [Google Scholar] [CrossRef]
- Hetland, M.A.K.; Hawkey, J.; Bernhoff, E.; Bakksjø, R.-J.; Kaspersen, H.; Rettedal, S.I.; Sundsfjord, A.; Holt, K.E.; Löhr, I.H. Within–patient and global evolutionary dynamics of Klebsiella pneumoniae ST17. Microb. Genom. 2023, 9, 001005. [Google Scholar] [CrossRef]
- Ravin, N.; Lane, D. Partition of the Linear Plasmid N15: Interactions of N15 Partition Functions with the sop Locus of the F Plasmid. J. Bacteriol. 1999, 181, 6898–6906. [Google Scholar] [CrossRef]
- Zinke, M.; Schröder, G.F.; Lange, A. Major tail proteins of bacteriophages of the order Caudovirales. J. Biol. Chem. 2022, 298, 101472. [Google Scholar] [CrossRef]
- Lo, C.-Y.; Gao, Y. DNA Helicase–Polymerase Coupling in Bacteriophage DNA Replication. Viruses 2021, 13, 1739. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lewis, D.E.A.; Adhya, S. The Developmental Switch in Bacteriophage λ: A Critical Role of the Cro Protein. J. Mol. Biol. 2018, 430, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2019; Volume 103, pp. 33–70. ISBN 978-0-12-817722-8. [Google Scholar]
- Jiang, S.; Chen, C.; Huang, W.; He, Y.; Du, X.; Wang, Y.; Ou, H.; Deng, Z.; Xu, C.; Jiang, L.; et al. A widespread phage-encoded kinase enables evasion of multiple host antiphage defence systems. Nat. Microbiol. 2024, 9, 3226–3239. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.B.; Crosti, G.; Dwivedi, B.; McDaniel, L.D.; Varsani, A.; Suttle, C.A.; Weinbauer, M.G.; Sandaa, R.-A.; Breitbart, M. Development of phoH as a Novel Signature Gene for Assessing Marine Phage Diversity. Appl. Environ. Microbiol. 2011, 77, 7730–7739. [Google Scholar] [CrossRef]
- Groth, A.C.; Calos, M.P. Phage Integrases: Biology and Applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef]
- Deneke, J.; Ziegelin, G.; Lurz, R.; Lanka, E. The protelomerase of temperate Escherichia coli phage N15 has cleaving-joining activity. Proc. Natl. Acad. Sci. USA 2000, 97, 7721–7726. [Google Scholar] [CrossRef]
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef]
- Logan, L.K.; Weinstein, R.A. The Epidemiology of Carbapenem-Resistant Enterobacteriaceae: The Impact and Evolution of a Global Menace. J. Infect. Dis. 2017, 215, S28–S36. [Google Scholar] [CrossRef]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a New Metallo-β-Lactamase Gene, bla NDM-1, and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef]
- Sreekumaran, S.; Priya, V.K.; Premnath, M.; Prathiush, P.R.; Anisha, M.N.; Mathew, J.; Jayachandran, K.; Radhakrishnan, E.K. Novel in-genome based analysis demonstrates the evolution of OmpK37, antimicrobial resistance gene from a potentially pathogenic pandrug resistant Klebsiella pneumoniae MS1 isolated from healthy broiler feces. Sci. Total Environ. 2024, 930, 172713. [Google Scholar] [CrossRef]
- Di Pilato, V.; Henrici De Angelis, L.; Aiezza, N.; Baccani, I.; Niccolai, C.; Parisio, E.M.; Giordano, C.; Camarlinghi, G.; Barnini, S.; Forni, S.; et al. Resistome and virulome accretion in an NDM-1-producing ST147 sublineage of Klebsiella pneumoniae associated with an outbreak in Tuscany, Italy: A genotypic and phenotypic characterisation. Lancet Microbe 2022, 3, e224–e234. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Alhejaili, A.Y.; Alkherd, U.H.; Milner, M.; Zhou, G.; Alzahrani, D.; Banzhaf, M.; Alzaidi, A.A.; Rajeh, A.A.; Al-Otaiby, M.A.; et al. The dissemination of multidrug-resistant and hypervirulent Klebsiella pneumoniae clones across the Kingdom of Saudi Arabia. Emerg. Microbes Infect. 2024, 13, 2427793. [Google Scholar] [CrossRef]
- Sonnevend, Á.; Abdulrazzaq, N.; Ghazawi, A.; Thomsen, J.; Bharathan, G.; Makszin, L.; Rizvi, T.A.; Pál, T. The first nationwide surveillance of carbapenem-resistant Enterobacterales in the United Arab Emirates—Increased association of Klebsiella pneumoniae CC14 clone with Emirati patients. Int. J. Infect. Dis. 2022, 120, 103–112. [Google Scholar] [CrossRef]
- Idrees, E.K.; Aldriwesh, M.G.; Alkhulaifi, M.M.; Alghoribi, M.F. Systematic review of multidrug-resistant Klebsiella pneumoniae in the Arabian Peninsula: Molecular epidemiology and resistance patterns. Front. Microbiol. 2025, 16, 1489317. [Google Scholar] [CrossRef] [PubMed]
- Kareem, S.M.; Al-Kadmy, I.M.S.; Kazaal, S.S.; Mohammed Ali, A.N.; Aziz, S.N.; Makharita, R.R.; Algammal, A.M.; Al-Rejaie, S.; Behl, T.; Batiha, G.E.-S.; et al. Detection of gyrA and parC Mutations and Prevalence of Plasmid-Mediated Quinolone Resistance Genes in Klebsiella pneumoniae. Infect. Drug Resist. 2021, 14, 555–563. [Google Scholar] [CrossRef]
- Doi, Y.; Wachino, J.; Arakawa, Y. Aminoglycoside Resistance. Infect. Dis. Clin. North Am. 2016, 30, 523–537. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Yamasaki, S.; Nakashima, R.; Zwama, M.; Hayashi-Nishino, M. Function and Inhibitory Mechanisms of Multidrug Efflux Pumps. Front. Microbiol. 2021, 12, 737288. [Google Scholar] [CrossRef]
- Huovinen, P.; Sundström, L.; Swedberg, G.; Sköld, O. Trimethoprim and Sulfonamide Resistance. Antimicrob. Agents Chemother. 1995, 39, 279–289. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kastoris, A.C.; Kapaskelis, A.M.; Karageorgopoulos, D.E. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum β-lactamase producing, Enterobacteriaceae infections: A systematic review. Lancet Infect. Dis. 2010, 10, 43–50. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed]
- Cervinkova, D.; Babak, V.; Marosevic, D.; Kubikova, I.; Jaglic, Z. The Role of the qacA Gene in Mediating Resistance to Quaternary Ammonium Compounds. Microbial. Drug Resistance 2013, 19, 160–167. [Google Scholar] [CrossRef]
- Rendón, M.A.; Saldaña, Z.; Erdem, A.L.; Monteiro-Neto, V.; Vázquez, A.; Kaper, J.B.; Puente, J.L.; Girón, J.A. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc. Natl. Acad. Sci. USA 2007, 104, 10637–10642. [Google Scholar] [CrossRef] [PubMed]
- Bachman, M.A.; Oyler, J.E.; Burns, S.H.; Caza, M.; Lépine, F.; Dozois, C.M.; Weiser, J.N. Klebsiella pneumoniae Yersiniabactin Promotes Respiratory Tract Infection Through Evasion of Lipocalin 2. Infect. Immun. 2011, 79, 3309–3316. [Google Scholar] [CrossRef]
- Smani, Y.; Fàbrega, A.; Roca, I.; Sánchez-Encinales, V.; Vila, J.; Pachón, J. Role of OmpA in the Multidrug Resistance Phenotype of Acinetobacter Baumannii. Antimicrob. Agents Chemother. 2014, 58, 1806–1808. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Seiffert, S.N.; Schwendener, S.; Perreten, V.; Endimiani, A. Differentiation of IncL and IncM Plasmids Associated with the Spread of Clinically Relevant Antimicrobial Resistance. PLoS ONE 2015, 10, e0123063. [Google Scholar] [CrossRef]
- Cascales, E.; Buchanan, S.K.; Duché, D.; Kleanthous, C.; Lloubès, R.; Postle, K.; Riley, M.; Slatin, S.; Cavard, D. Colicin Biology. Microbiol. Mol. Biol. Rev. 2007, 71, 158–229. [Google Scholar] [CrossRef]
- Hertwig, S.; Klein, I.; Schmidt, V.; Beck, S.; Hammerl, J.A.; Appel, B. Sequence Analysis of the Genome of the Temperate Yersinia Enterocolitica Phage PY54. J. Mol. Biol. 2003, 331, 605–622. [Google Scholar] [CrossRef]
- Kelly, A.; Arrowsmith, T.J.; Went, S.C.; Blower, T.R. Toxin–antitoxin systems as mediators of phage defence and the implications for abortive infection. Curr. Opin. Microbiol. 2023, 73, 102293. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Kostyuchenko, V.A.; Efimov, A.V.; Kurochkina, L.P.; Sykilinda, N.N.; Robben, J.; Volckaert, G.; Hoenger, A.; Chipman, P.R.; Battisti, A.J.; et al. A Three-dimensional Cryo-electron Microscopy Structure of the Bacteriophage ϕKZ Head. J. Mol. Biol. 2005, 352, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Cresawn, S.G.; Hendrix, R.W. Comparative genomics of the mycobacteriophages: Insights into bacteriophage evolution. Res. Microbiol. 2008, 159, 332–339. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.D.; Smith, B.T.; Godoy, V.G.; Walker, G.C. THE SOS RESPONSE: Recent Insights into umuDC -Dependent Mutagenesis and DNA Damage Tolerance. Annu. Rev. Genet. 2000, 34, 479–497. [Google Scholar] [CrossRef]
- Gutierrez, A.; Laureti, L.; Crussard, S.; Abida, H.; Rodríguez-Rojas, A.; Blázquez, J.; Baharoglu, Z.; Mazel, D.; Darfeuille, F.; Vogel, J.; et al. β-lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat. Commun. 2013, 4, 1610. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).