
A Genomic Surveillance Circuit for Emerging Viral Pathogens

 , , , ,
, , , ,  , , , , ,
, , , , ,  , , and
, , and
Abstract
:1. Introduction
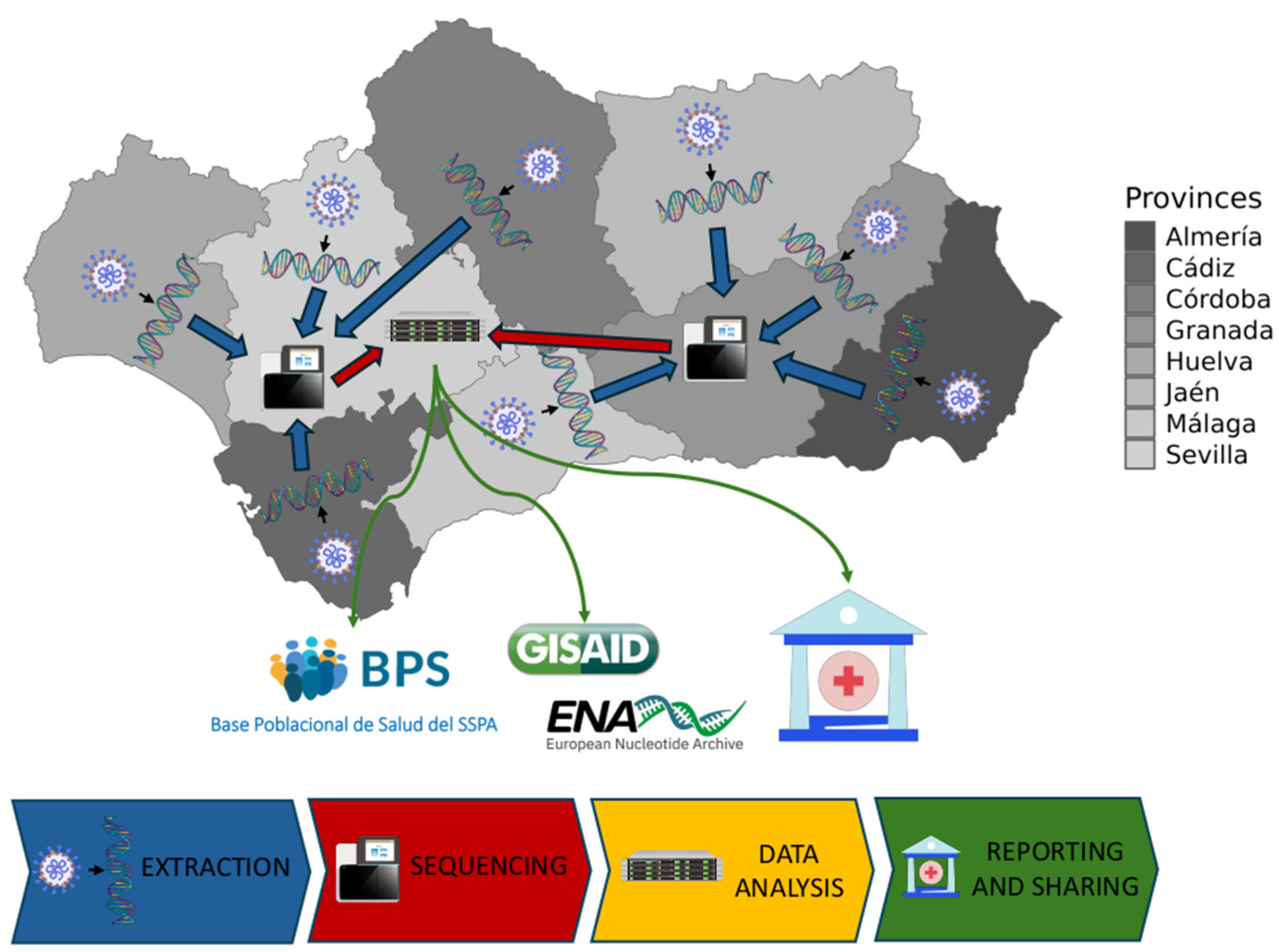
2. Materials and Methods
2.1. Design and Patient Selection
2.2. SARS-CoV-2 Genome Sequencing
2.3. Illumina Sequencing Data Processing Workflow
2.4. Nanopore Sequencing Data Processing Workflow
2.5. Phylogenetic Analysis
2.6. Resolution and Performance of the Andalusian Surveillance Circuit
3. Results
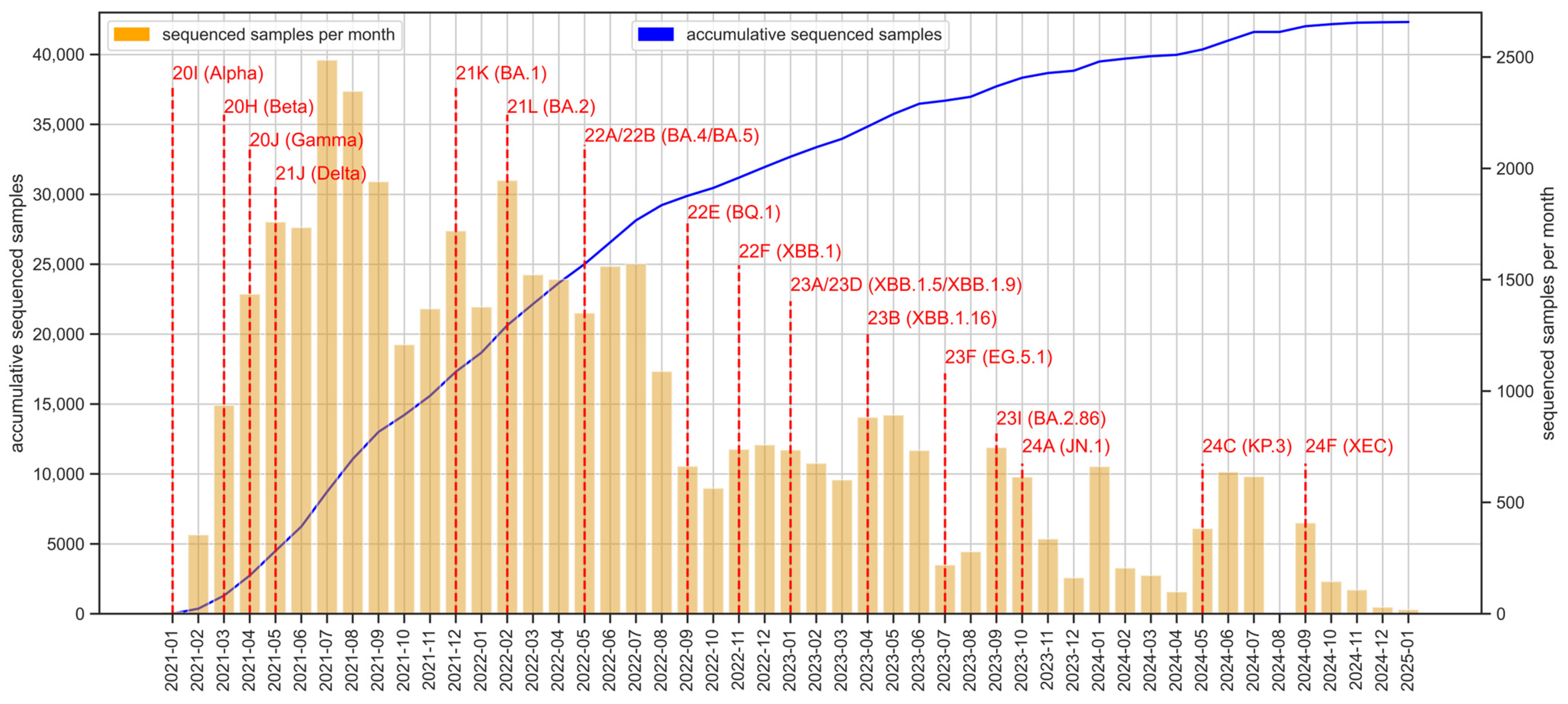
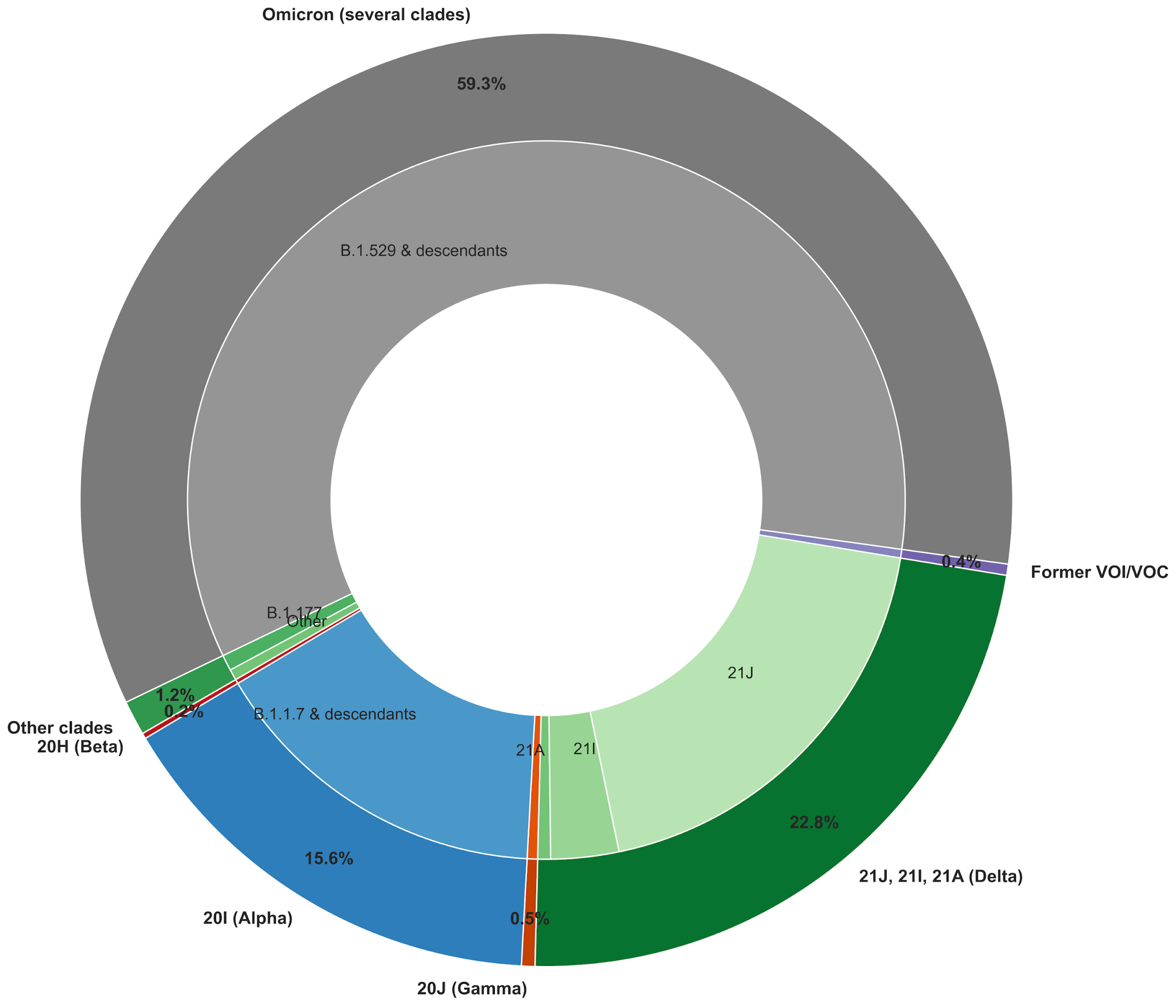
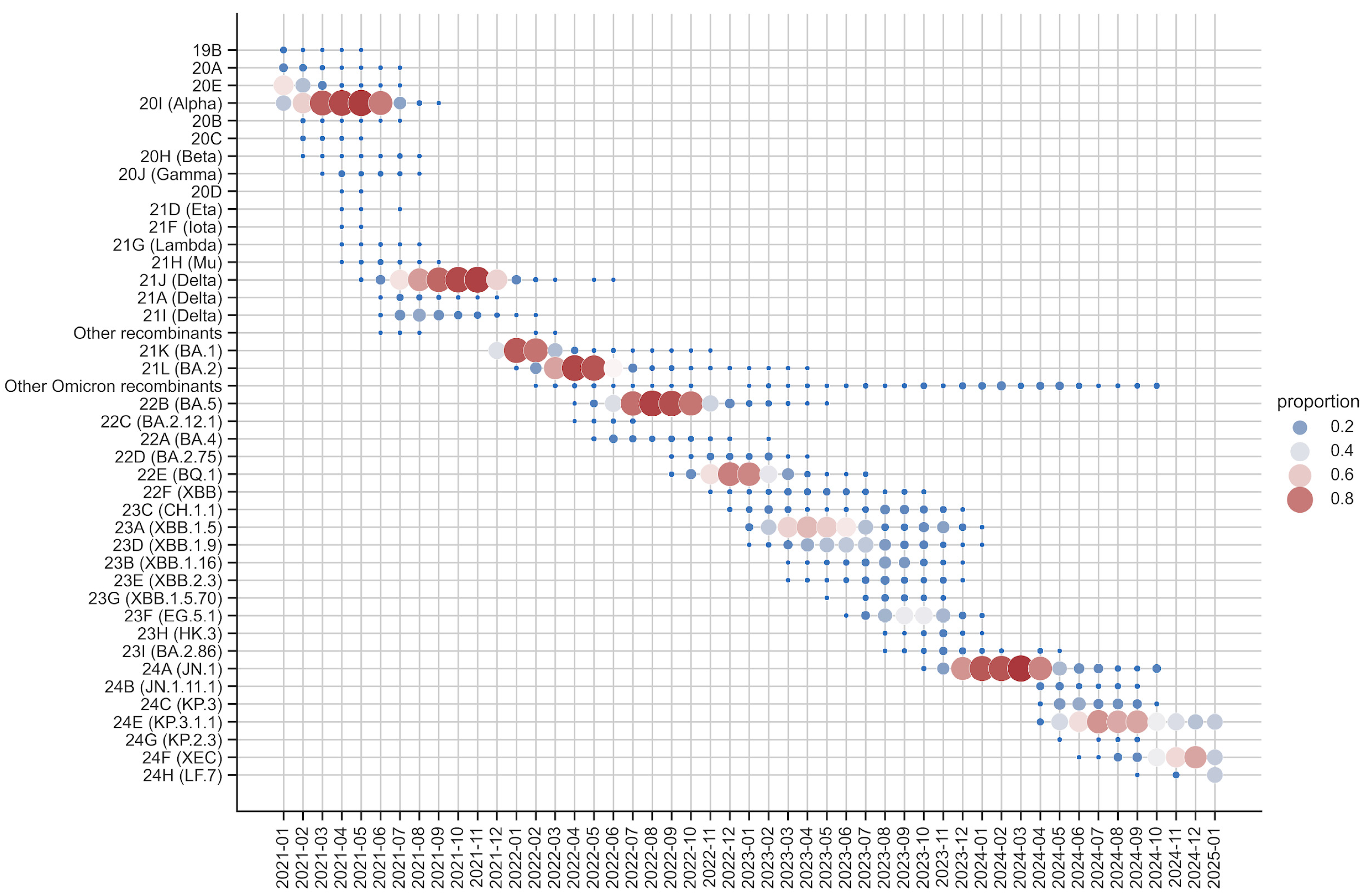
3.1. Sequencing Effort over the 2021–2025 Period
3.2. Nextstrain Local Server
3.3. Impact of Genomic Surveillance on Public Health Interventions in Andalusia
3.4. Performance Evaluation Through Quality Control Assessments (QCAs)
3.5. Use Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| VOI | Variants of interest |
| VOC | Variants of concern |
| VUM | Variants under monitoring |
| EHR | Electronic health record |
| BPS | Base Poblacional de Salud |
| GPU | Graphics processing unit |
| RWE | Real-world Evidence |
| WGS | Whole-genome sequencing |
References
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.; Sales, F.C.; Hawryluk, I.; McCrone, J.T. Genomics and epidemiology of the P. 1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á. Assessing transmissibility of SARS-CoV-2 lineage, B. 1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Beyer, D.K.; Forero, A. Mechanisms of Antiviral Immune Evasion of SARS-CoV-2. J. Mol. Biol. 2022, 434, 167265. [Google Scholar] [CrossRef]
- Cyranoski, D. Alarming COVID variants show vital role of genomic surveillance. Nature 2021, 589, 337–338. [Google Scholar] [CrossRef]
- WHO. SARS-CoV-2 Variants of Concern and Variants of Interest. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 12 January 2025).
- Porter, A.F.; Sherry, N.; Andersson, P.; Johnson, S.A.; Duchene, S.; Howden, B.P. New rules for genomics-informed COVID-19 responses-Lessons learned from the first waves of the Omicron variant in Australia. PLoS Genet. 2022, 18, e1010415. [Google Scholar] [CrossRef]
- Sequencing of the SARS-CoV-2 Virus Genome for the Monitoring and Management of the COVID-19 Epidemic in Andalusia and the Rapid Generation of Prognostic and Response to Treatment Biomarkers. Available online: https://www.clinbioinfosspa.es/projects/covseq/indexEng.html (accessed on 12 January 2025).
- SARS-CoV-2 Whole Genome Sequencing Circuit in Andalusia. Available online: https://www.clinbioinfosspa.es/COVID_circuit/ (accessed on 12 January 2025).
- Vázquez-Morón, S.; Iglesias-Caballero, M.; Lepe, J.A.; Garcia, F.; Melón, S.; Marimon, J.M.; de Viedma, D.G.; Folgueira, M.D.; Galán, J.C.; López-Causapé, C.; et al. Enhancing SARS-CoV-2 Surveillance through Regular Genomic Sequencing in Spain: The RELECOV Network. Int. J. Mol. Sci. 2023, 24, 8573. [Google Scholar] [CrossRef] [PubMed]
- Dopazo, J.; Maya-Miles, D.; García, F.; Lorusso, N.; Calleja, M.Á.; Pareja, M.J.; López-Miranda, J.; Rodríguez-Baño, J.; Padillo, J.; Túnez, I. Implementing Personalized Medicine in COVID-19 in Andalusia: An Opportunity to Transform the Healthcare System. J. Pers. Med. 2021, 11, 475. [Google Scholar] [CrossRef] [PubMed]
- Muñoyerro-Muñiz, D.; Goicoechea-Salazar, J.; García-León, F.; Laguna-Tellez, A.; Larrocha-Mata, D.; Cardero-Rivas, M. Health record linkage: Andalusian health population database. Gac. Sanit. 2019, 34, 105–113. [Google Scholar] [CrossRef]
- ISCIII. Integration of Genome Sequencing in the SARS-CoV-2 Surveillance. Available online: https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Integracion_de_la_secuenciacion_genomica-en_la_vigilancia_del_SARS-CoV-2.pdf (accessed on 13 February 2025).
- ARTIC Network. Available online: https://community.artic.network/ (accessed on 13 February 2025).
- ARTIC Network. V3 Primer Availability. Available online: https://community.artic.network/t/v3-primer-availability/123 (accessed on 13 February 2025).
- ARTIC Network. SARS-CoV-2 Version 4 Scheme Release. Available online: https://community.artic.network/t/sars-cov-2-version-4-scheme-release/312 (accessed on 13 February 2025).
- ARTIC Network. SARS-CoV-2 V4.1 Update for Omicron Variant. Available online: https://community.artic.network/t/sars-cov-2-v4-1-update-for-omicron-variant/342 (accessed on 13 February 2025).
- ARTIC Network. SARS-CoV-2 Version 5.3.2 Scheme Release. Available online: https://community.artic.network/t/sars-cov-2-version-5-3-2-scheme-release/462 (accessed on 13 February 2025).
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Monzón, S.; Varona, S.; Espinosa-Carrasco, J.; Garcia, M.U.; Nf-Core Bot; Ewels, P. nf-core/viralrecon: Nf-core/viralrecon v2.6.0—Rhodium Raccoon. Zenodo 2023, 7764938. Available online: https://zenodo.org/records/7764938 (accessed on 13 February 2025).
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Li, B.; Huang, H.-L.; Chen, H.-D.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; Goes De Jesus, J.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Broad Institute. Picard Toolkit; Broad Institute, GitHub Repository. 2018. Available online: http://broadinstitute.github.io/picard/ (accessed on 13 February 2025).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- ARTIC Network. The ARTIC Field Bioinformatics Pipeline. Available online: https://github.com/artic-network/fieldbioinformatics (accessed on 21 February 2025).
- Nanopolish. A Software Package for Signal-Level Analysis of Oxford Nanopore Sequencing Data. Available online: https://nanopolish.readthedocs.io/en/latest/index.html (accessed on 5 April 2025).
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences; University of Utah: Salt Lake City, UT, USA, 1986. [Google Scholar]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- EPI_SET_250402xc. High-Quality SARS-CoV-2 Genomes Deposited in GISAID from Spain, Excluding Andalusia Region. Available online: https://doi.org/10.55876/gis8.250402xc (accessed on 5 April 2025).
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Cantoni, D.; Scott, S.; Logan, N.; Ashraf, S.; et al. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4, and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Tamura, T.; Irie, T.; Deguchi, S.; Yajima, H.; Tsuda, M.; Nasser, H.; Mizuma, K.; Plianchaisuk, A.; Suzuki, S.; Uriu, K.; et al. Virological characteristics of the SARS-CoV-2 Omicron XBB.1.5 variant. Nat. Commun. 2024, 15, 1176. [Google Scholar] [CrossRef]
- Wang, Q.; Mellis, I.A.; Ho, J.; Bowen, A.; Kowalski-Dobson, T.; Valdez, R.; Katsamba, P.S.; Wu, M.; Lee, C.; Shapiro, L.; et al. Recurrent SARS-CoV-2 spike mutations confer growth advantages to select JN.1 sublineages. Emerg. Microbes Infect. 2024, 13, 2402880. [Google Scholar] [CrossRef]
- The SARS-CoV-2 Genomic Surveillance Circuit of Andalusia, Nextstrain Server. Available online: https://nextstrain.clinbioinfosspa.es/SARS-COV-2-2021-2025 (accessed on 14 February 2025).
- Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health; WHO: Geneva, Switzerland. 2021, pp. 1–64. Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 5 April 2025).
- Global Genomic Surveillance Strategy for Pathogens with Pandemic and Epidemic Potential, 2022–2032. Available online: https://www.who.int/initiatives/genomic-surveillance-strategy (accessed on 5 April 2025).
- Gong, Y.N.; Kuo, N.Y.; Yeh, T.S.; Shih, S.R.; Chen, G.W. Genomic Surveillance of SARS-CoV-2 in Taiwan: A Perspective on Evolutionary Data Interpretation and Sequencing Issues. Biomed. J. 2024. Online ahead of print. [Google Scholar] [CrossRef]
- Salud Organiza un Cribado en La Línea y Algeciras y Reclama al Gobierno Mayor Control en el Tránsito con Gibraltar. Available online: https://www.juntadeandalucia.es/presidencia/portavoz/salud/157252/Covid/coronavirus/pandemia/cribado/CampodeGibraltrar/Algeciras/LaLineadelaConcepcion/Brexit/Gibraltar/Cadiz/Andalucia/JuntadeAndalucia/GobiernodeAndalucia (accessed on 5 April 2025).
- Loucera, C.; Perez-Florido, J.; Casimiro-Soriguer, C.S.; Ortuño, F.M.; Carmona, R.; Bostelmann, G.; Martínez-González, L.J.; Muñoyerro-Muñiz, D.; Villegas, R.; Rodriguez-Baño, J.; et al. Assessing the impact of SARS-CoV-2 lineages and mutations on patient survival. Viruses 2022, 14, 1893. [Google Scholar] [CrossRef] [PubMed]
- Loucera, C.; Peña-Chilet, M.; Esteban-Medina, M.; Muñoyerro-Muñiz, D.; Villegas, R.; Lopez-Miranda, J.; Rodriguez-Baño, J.; Túnez, I.; Bouillon, R.; Dopazo, J. Real world evidence of calcifediol or vitamin D prescription and mortality rate of COVID-19 in a retrospective cohort of hospitalized Andalusian patients. Sci. Rep. 2021, 11, 23380. [Google Scholar] [CrossRef]
- Loucera-Muñecas, C.; Canal-Rivero, M.; Ruiz-Veguilla, M.; Carmona, R.; Bostelmann, G.; Garrido-Torres, N.; Dopazo, J.; Crespo-Facorro, B. Aripiprazole as protector against COVID-19 mortality. Sci. Rep. 2024, 14, 12362. [Google Scholar] [CrossRef]
- Loucera, C.; Carmona, R.; Esteban-Medina, M.; Bostelmann, G.; Muñoyerro-Muñiz, D.; Villegas, R.; Peña-Chilet, M.; Dopazo, J. Real-world evidence with a retrospective cohort of 15,968 COVID-19 hospitalized patients suggests 21 new effective treatments. Virol. J. 2023, 20, 226. [Google Scholar] [CrossRef] [PubMed]
- Perez-Florido, J.; Casimiro-Soriguer, C.S.; Ortuño, F.; Fernandez-Rueda, J.L.; Aguado, A.; Lara, M.; Riazzo, C.; Rodriguez-Iglesias, M.A.; Camacho-Martinez, P.; Merino-Diaz, L.; et al. Detection of high levels of co-infection and the emergence of novel SARS-CoV-2 Delta-Omicron and Omicron-Omicron recombinants in the epidemiological surveillance of Andalusia. Int. J. Mol. Sci. 2023, 24, 2419. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Blanco, L.; de Salazar, A.; Fuentes, A.; Viñuela, L.; Perez-Florido, J.; Dopazo, J.; García, F. Evaluation of a combined detection of SARS-CoV-2 and its variants using real-time allele-specific PCR strategy: An advantage for clinical practice. Epidemiol. Infect. 2023, 151, e201. [Google Scholar] [CrossRef]
- Ortuño, F.M.; Loucera, C.; Casimiro-Soriguer, C.S.; Lepe, J.A.; Camacho-Martinez, P.; Merino-Diaz, L.; de Salazar, A.; Chueca, N.; García, F.; Perez-Florido, J.; et al. Highly accurate whole-genome imputation of SARS-CoV-2 from partial or low-quality sequences. GigaScience 2021, 10, giab078. [Google Scholar]
- COVID-19 Genomics UK (COG-UK) Consortium. Available online: https://webarchive.nationalarchives.gov.uk/ukgwa/20230505083137/https://www.cogconsortium.uk/ (accessed on 5 April 2025).
- COVID-19 Genomics Consortium Denmark. Available online: https://www.covid19genomics.dk/home (accessed on 5 April 2025).
- Consortium EMERGEN. Available online: https://www.santepubliquefrance.fr/dossiers/coronavirus-covid-19/consortium-emergen (accessed on 5 April 2025).
- Wegner, F.; Cabrera-Gil, B.; Tanguy, A.; Beckmann, C.; Beerenwinkel, N.; Bertelli, C.; Carrara, M.; Cerutti, L.; Chen, C.; Cordey, S.; et al. How Much Should We Sequence? An Analysis of the Swiss SARS-CoV-2 Surveillance Effort. Microbiol. Spectr. 2024, 12, e0362823. [Google Scholar] [CrossRef] [PubMed]
- Genetic Diversity of the Novel Coronavirus SARS-CoV-2 (COVID-19) in Portugal. Available online: https://insaflu.insa.pt/covid19/ (accessed on 5 April 2025).
- Frank, O.; Balboa, D.A.; Novatchkova, M.; Özkan, E.; Strobl, M.M.; Yelagandula, R.; Albanese, T.G.; Endler, L.; Amman, F.; Felsenstein, V.; et al. Genomic Surveillance of SARS-CoV-2 Evolution by a Centralised Pipeline and Weekly Focused Sequencing, Austria, January 2021 to March 2023. Eurosurveillance 2024, 29, 2300542. [Google Scholar] [CrossRef]
- The Whole Genome Sequencing Surveillance Circuit of Andalusia. Available online: https://www.clinbioinfosspa.es/surveillance_circuit/ (accessed on 14 February 2025).
- Casimiro-Soriguer, C.S.; Perez-Florido, J.; Fernandez-Rueda, J.L.; Pedrosa-Corral, I.; Guillot-Sulay, V.; Lorusso, N.; Martinez-Gonzalez, L.J.; Navarro-Marí, J.M.; Dopazo, J.; Sanbonmatsu-Gámez, S. Phylogenetic Analysis of the 2020 West Nile Virus (WNV) Outbreak in Andalusia (Spain). Viruses 2021, 13, 836. [Google Scholar] [CrossRef]
- Casimiro-Soriguer, C.S.; Perez-Florido, J.; Lara, M.; Camacho-Martinez, P.; Merino-Diaz, L.; Pupo-Ledo, I.; de Salazar, A.; Fuentes, A.; Viñuela, L.; Chueca, N.; et al. Molecular and phylogenetic characterization of the monkeypox outbreak in the South of Spain. Health Sci. Rep. 2024, 7, e1965. [Google Scholar] [CrossRef]
- Casimiro-Soriguer, C.S.; Pérez-Florido, J.; Robles, E.A.; Lara, M.; Aguado, A.; Rodríguez Iglesias, M.A.; Lepe, J.A.; García, F.; Pérez-Alegre, M.; Andújar, E.; et al. The Integrated Genomic Surveillance System of Andalusia (SIEGA) Provides a One Health Regional Resource Connected with the Clinic. Sci. Rep. 2024, 14, 19200. [Google Scholar] [CrossRef]
- Neves, A.; Willassen, N.P.; Hjerde, E.; Cuesta, I.; Martin, C.S.; Inno, H.; Pilvar, D.; Ng, K.; Salgado, D.; van Helden, J.; et al. ELIXIR CONVERGE WP9 community. A survey into the contribution of regional/national pathogen data platforms and on the resources needed to develop and maintain them. F1000Research 2024, 12, 1590. [Google Scholar] [CrossRef]
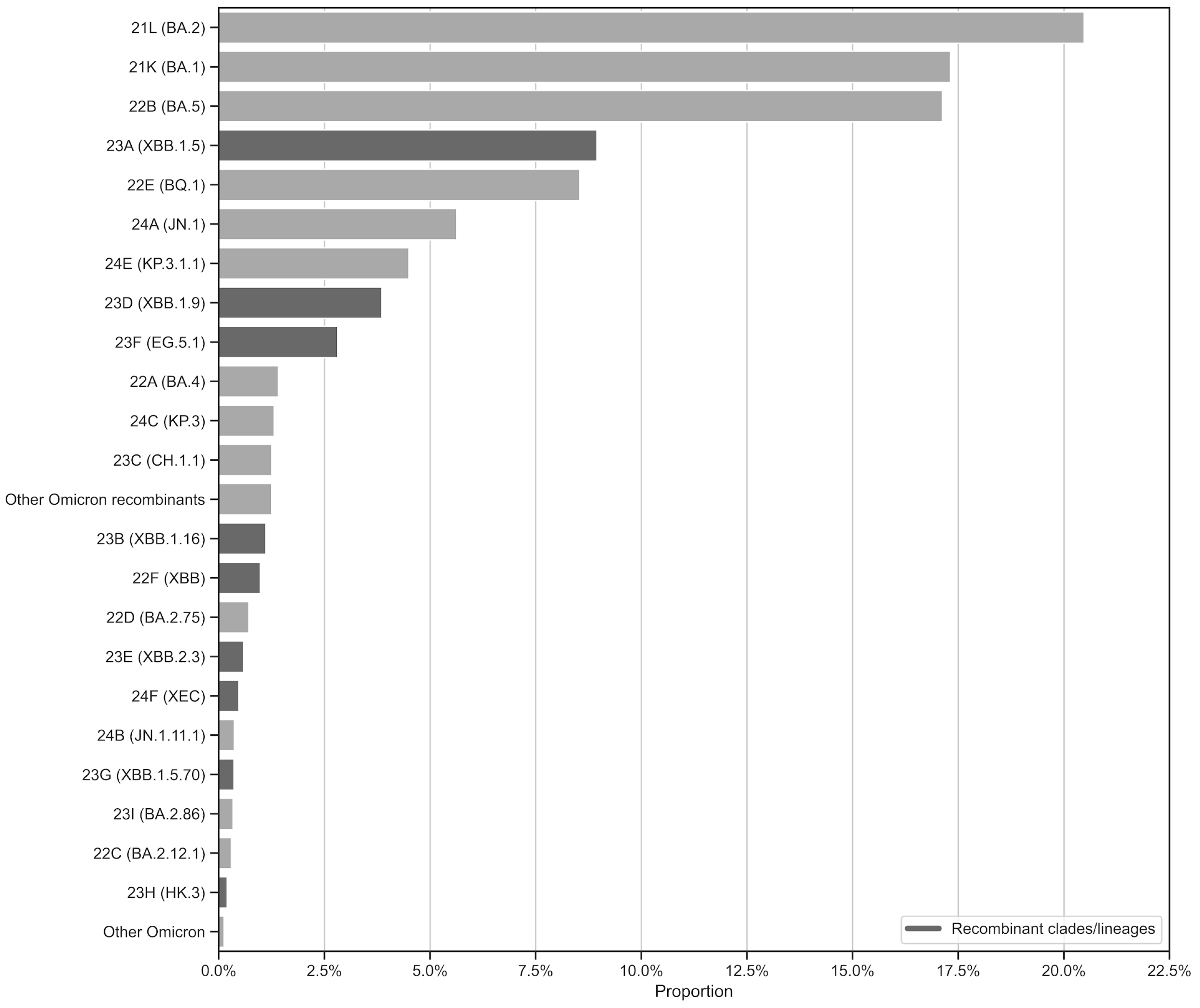



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Reference Clade | Reference Lineage | Clade: Number of Correct Results (%) | Lineage: Number of Correct Results (%) |
|---|---|---|---|---|
| QCA-01-2021 | 20I | B.1.1.7 | 2 (100) | 2 (100) |
| QCA-02-2021 | 20H | B.1.351 | 2 (100) | 2 (100) |
| QCA-03-2021 | 19B | A.28 | 2 (100) | 2 (100) |
| QCA-04-2021 | 21H | B.1.621 | 2 (100) | 2 (100) |
| QCA-05-2021 | 20J | P.1 | 2 (100) | 2 (100) |
| QCA-06-2021 | 21I | AY.9.2 | 2 (100) | 2 (100) |
| QCA-08-2021 | 21J | AY.94 | 2 (100) | 2 (100) |
| QCA-09-2021 | 21J | AY.94 | 2 (100) | 2 (100) |
| QCA-10-2021 | 21J | AY.43 | 2 (100) | 2 (100) |
| QCA-02-2024 | 23B | XBB.1.16 | 3 (100) | 3 (100) |
| QCA-03-2024 | 23A | XBB.1.5 | 2 (66.66) | 2 (66.66) |
| QCA-04-2024 | 24A | JN.1.59 | 3 (100) | 3 (100) |
| QCA-05-2024 | 23A | XBB.1.5 | 3 (100) | 3 (100) |
| QCA-07-2024 | 24A | JN.1 | 3 (100) | 3 (100) |
| QCA-10-2024 | 24A | JN.1 | 2 (66.66) | 1 (33.33) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casimiro-Soriguer, C.S.; Lara, M.; Aguado, A.; Loucera, C.; Ortuño, F.M.; Lorusso, N.; Navarro-Marí, J.M.; Sanbonmatsu-Gámez, S.; Camacho-Martinez, P.; Merino-Diaz, L.; et al. A Genomic Surveillance Circuit for Emerging Viral Pathogens. Microorganisms 2025, 13, 912. https://doi.org/10.3390/microorganisms13040912
Casimiro-Soriguer CS, Lara M, Aguado A, Loucera C, Ortuño FM, Lorusso N, Navarro-Marí JM, Sanbonmatsu-Gámez S, Camacho-Martinez P, Merino-Diaz L, et al. A Genomic Surveillance Circuit for Emerging Viral Pathogens. Microorganisms. 2025; 13(4):912. https://doi.org/10.3390/microorganisms13040912
Chicago/Turabian StyleCasimiro-Soriguer, Carlos S., Maria Lara, Andrea Aguado, Carlos Loucera, Francisco M. Ortuño, Nicola Lorusso, Jose M. Navarro-Marí, Sara Sanbonmatsu-Gámez, Pedro Camacho-Martinez, Laura Merino-Diaz, and et al. 2025. "A Genomic Surveillance Circuit for Emerging Viral Pathogens" Microorganisms 13, no. 4: 912. https://doi.org/10.3390/microorganisms13040912
APA StyleCasimiro-Soriguer, C. S., Lara, M., Aguado, A., Loucera, C., Ortuño, F. M., Lorusso, N., Navarro-Marí, J. M., Sanbonmatsu-Gámez, S., Camacho-Martinez, P., Merino-Diaz, L., de Salazar, A., Fuentes, A., The Andalusian COVID-19 Sequencing Initiative, Lepe, J. A., García, F., Dopazo, J., & Perez-Florido, J. (2025). A Genomic Surveillance Circuit for Emerging Viral Pathogens. Microorganisms, 13(4), 912. https://doi.org/10.3390/microorganisms13040912