
Identification and Characterization of Two Novel Members of the Family Eubacteriaceae, Anaerofustis butyriciformans sp. nov. and Pseudoramibacter faecis sp. nov., Isolated from Human Feces

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Treatment
2.2. Culture Media and Preservation
2.3. Morphological, Physiological and Biochemical Taxonomic Determinations
2.4. Phylogenetic and Genomic Analysis
3. Results
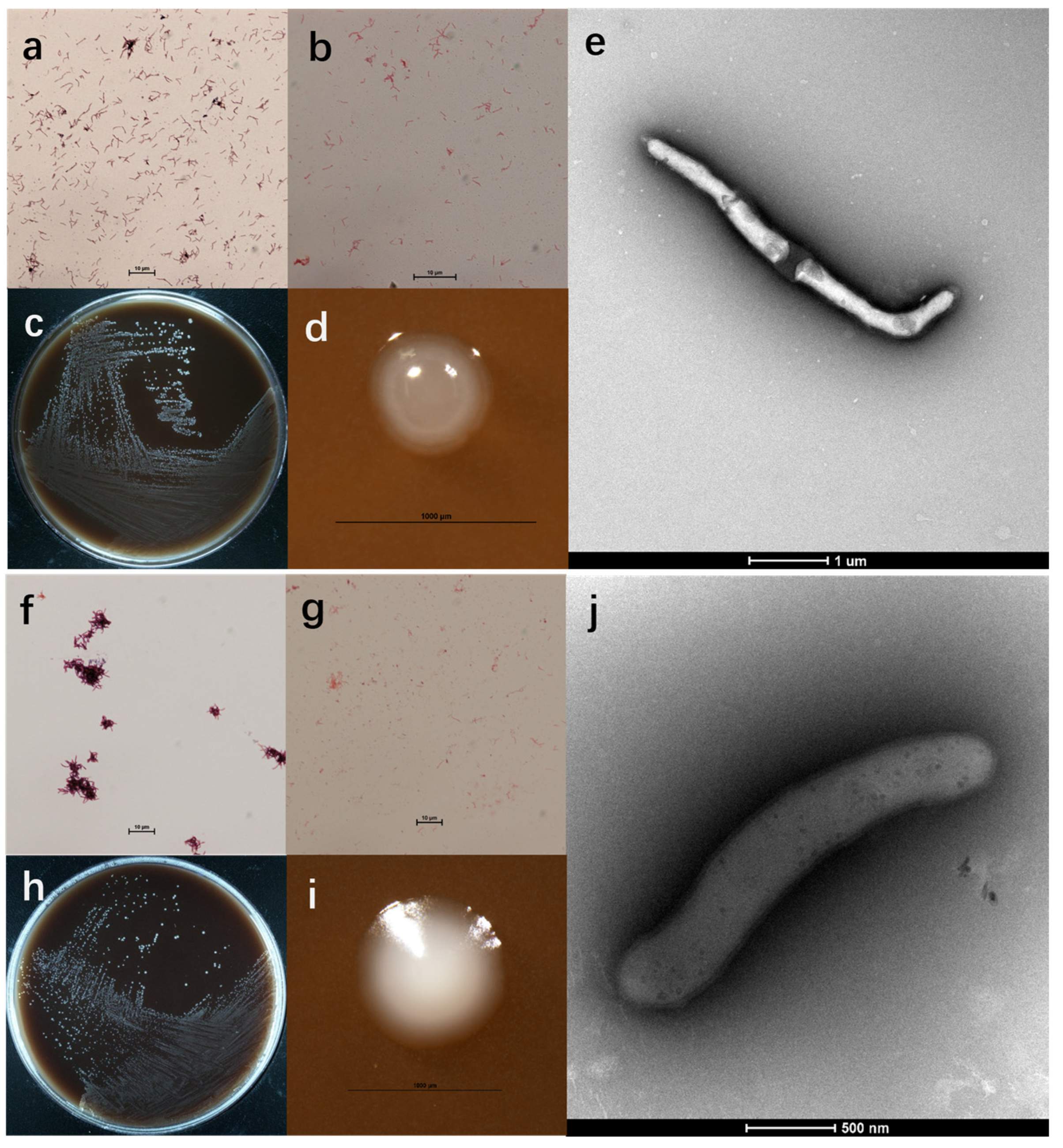
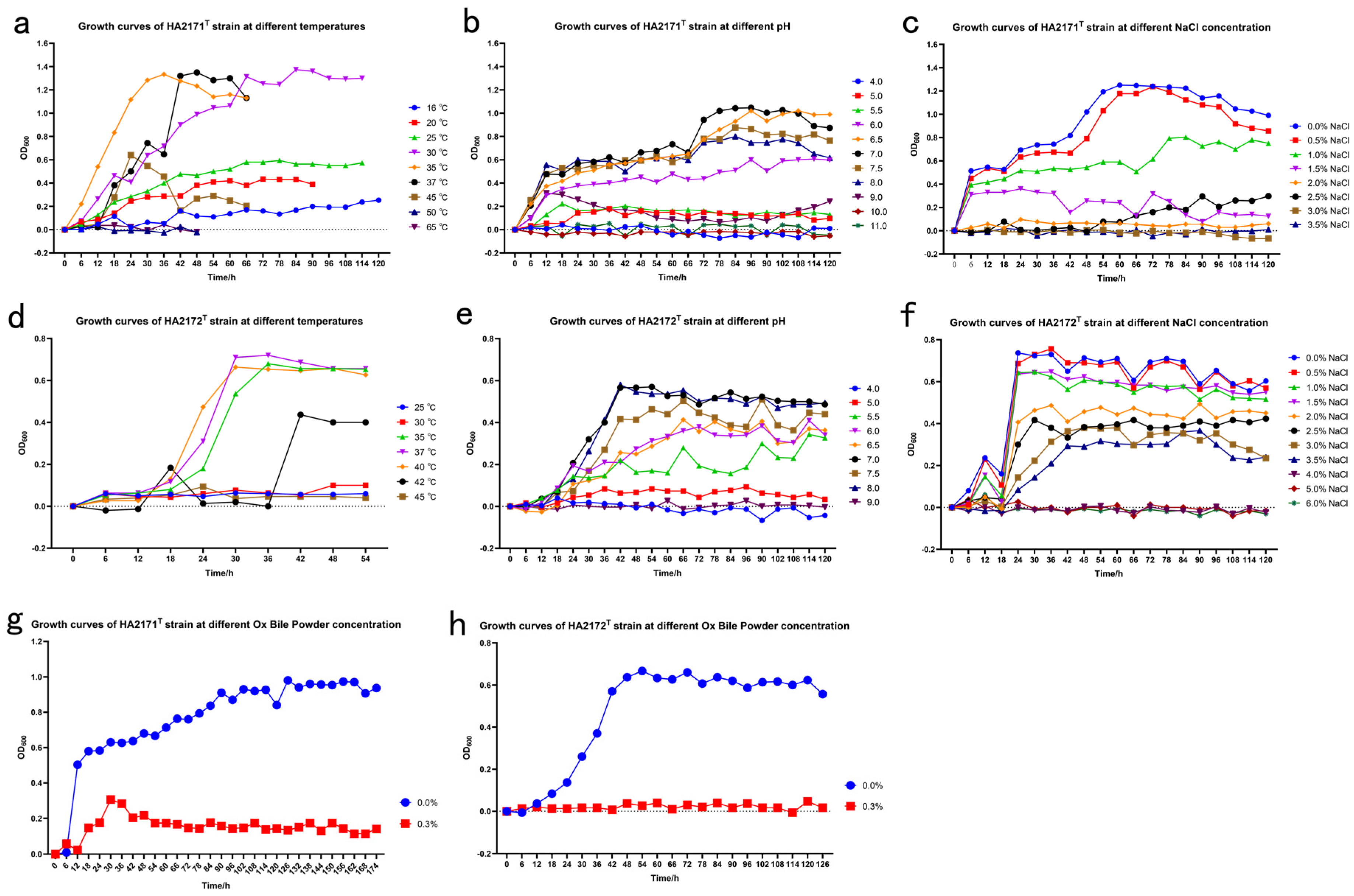
3.1. Morphological and Physiological Properties
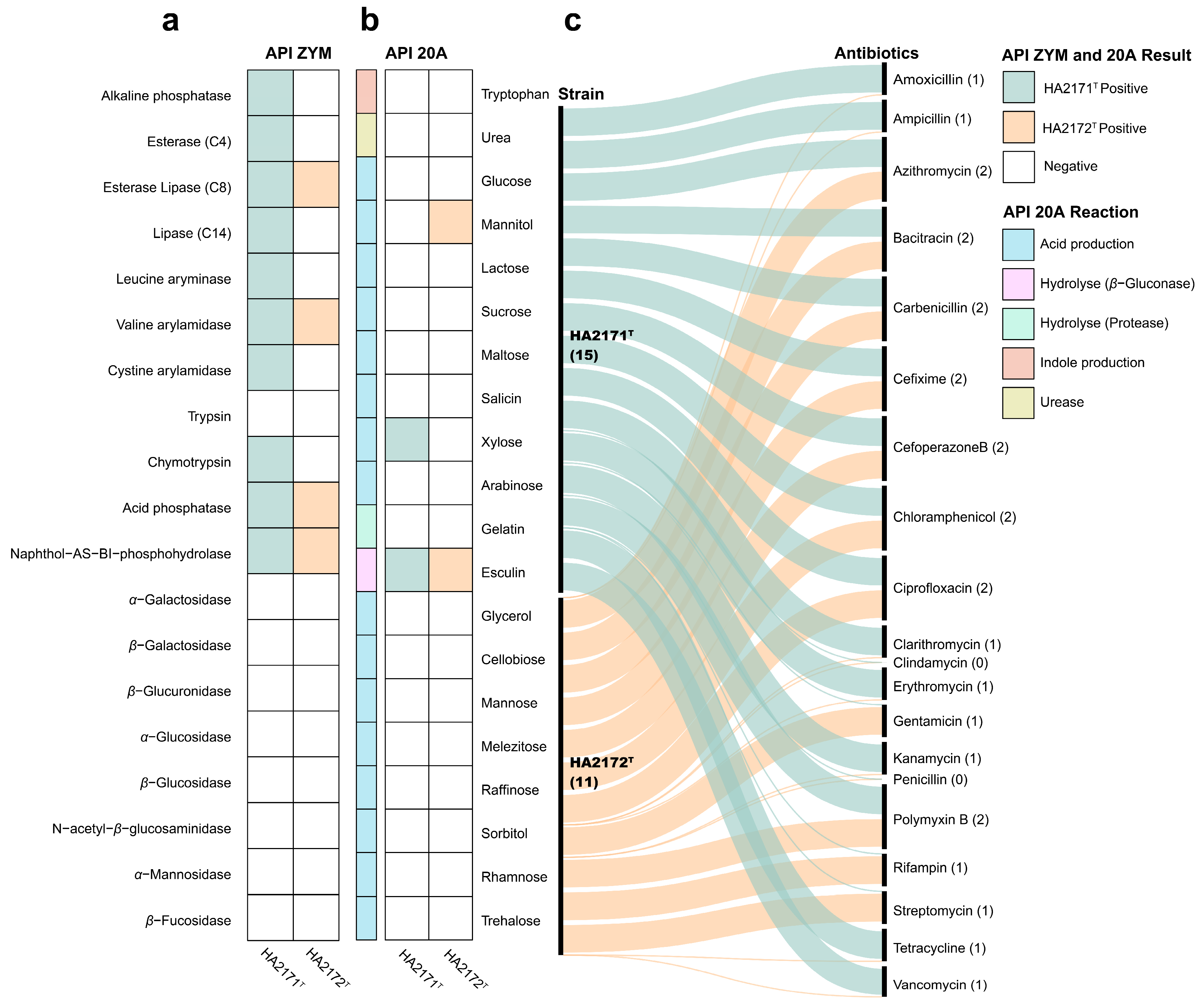
3.2. Chemotaxonomic Characteristics
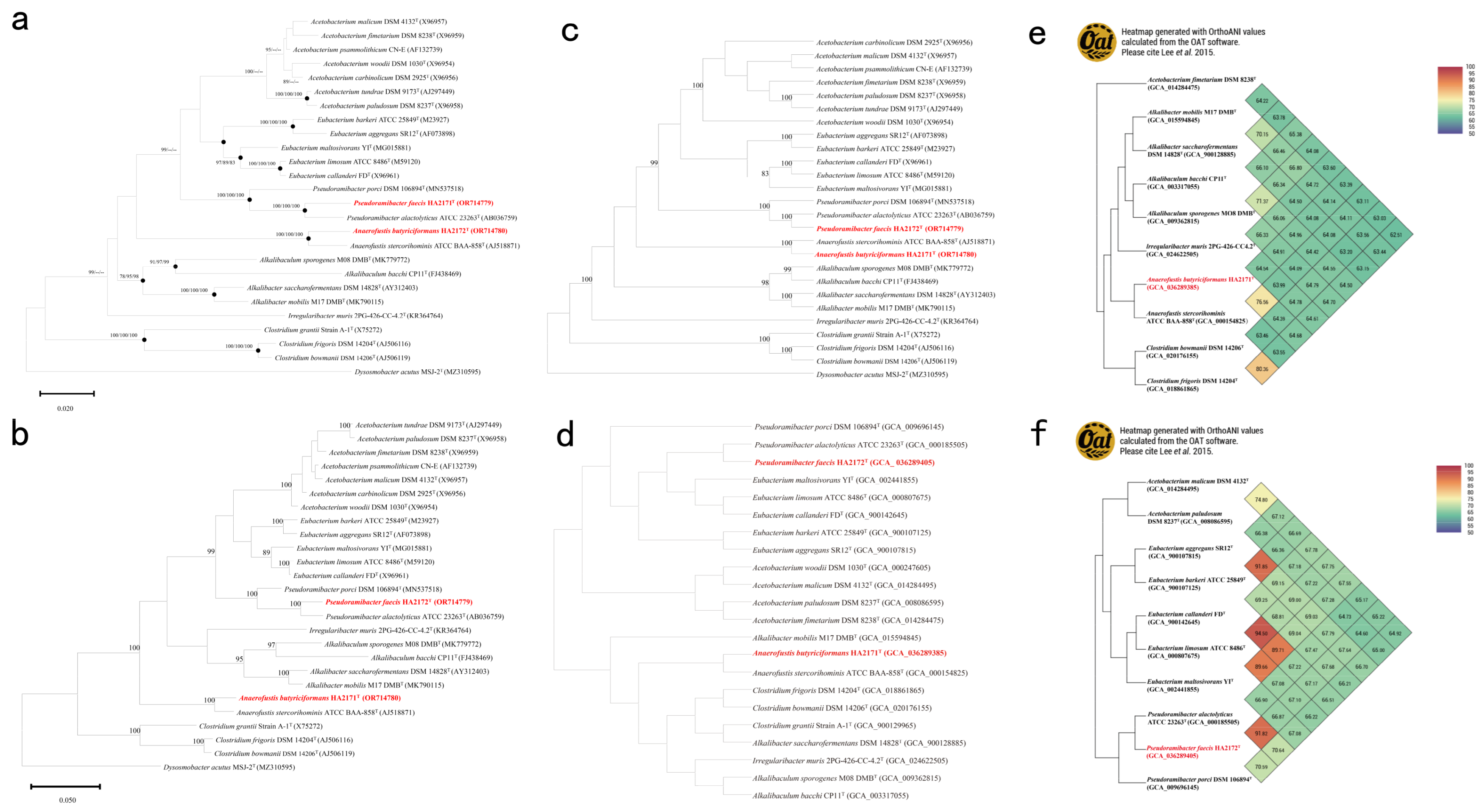
3.3. Phylogeny and Phylogenomic Features
3.4. In Silico Analysis of Biological Characterization and Gut-Associated Functional Potential
3.4.1. Taxonomic Differentiation
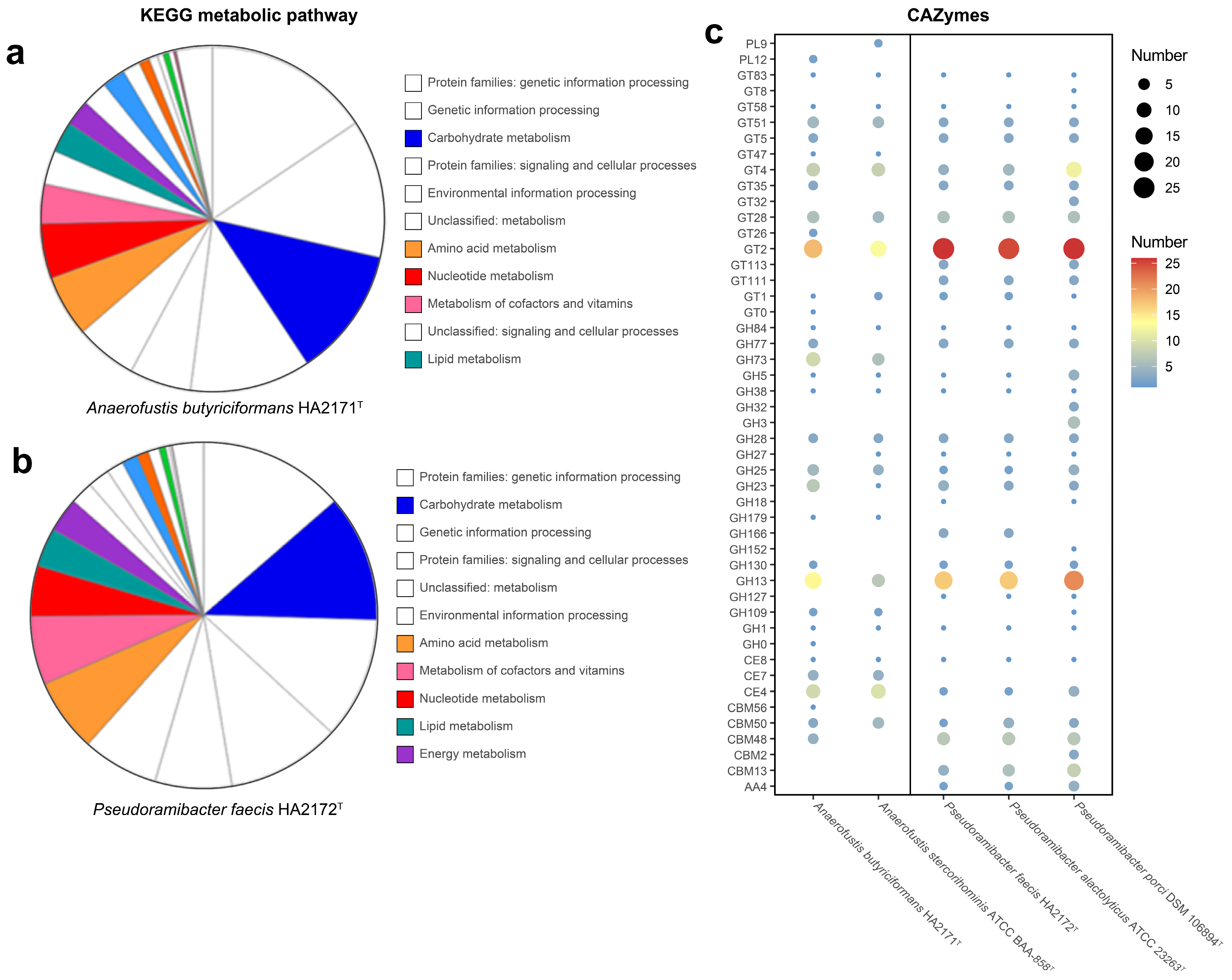
3.4.2. Metabolic Potential
3.4.3. Implications for Human Health
4. Discussion
5. Conclusions
6. Descriptions
6.1. Description of Anaerofustis butyriciformans sp. nov.
6.2. Description of Pseudoramibacter faecis sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Schleifer, K.H.; Whitman, W.B. Family II. Eubacteriaceae fam. nov. In Bergey Manual of Systematic Bacteriology, 2nd ed.; Vos, P.D., Garrity, G.M., Jones, D., Krieg, N.R., Ludwig, W., Rainey, F.A., Schleifer, K.H., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2009; Volume 3, pp. 865–875. [Google Scholar]
- Finegold, S.M.; Lawson, P.A.; Vaisanen, M.-L.; Molitoris, D.R.; Song, Y.; Liu, C.; Collins, M.D. Anaerofustis stercorihominis gen. nov., sp. nov., from human feces. Anaerobe 2004, 10, 41–45. [Google Scholar] [CrossRef]
- Wylensek, D.; Hitch, T.C.A.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernández, S.B.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 2020, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; Collins, M.D. Phylogenetic relationships of the genera Acetobacterium and Eubacterium sensu stricto and reclassification of Eubacterium alactolyticum as Pseudoramibacter alactolyticus gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 1996, 46, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Greses, S.; De Bernardini, N.; Treu, L.; Campanaro, S.; González-Fernández, C. Genome-centric metagenomics revealed the effect of pH on the microbiome involved in short-chain fatty acids and ethanol production. Bioresour. Technol. 2023, 377, 128920. [Google Scholar] [CrossRef]
- Karcher, N.; Pasolli, E.; Asnicar, F.; Huang, K.D.; Tett, A.; Manara, S.; Armanini, F.; Bain, D.; Duncan, S.H.; Louis, P.; et al. Analysis of 1321 Eubacterium rectale genomes from metagenomes uncovers complex phylogeographic population structure and subspecies functional adaptations. Genome Biol. 2020, 21, 138. [Google Scholar] [CrossRef]
- Lu, H.; Xu, X.; Fu, D.; Gu, Y.; Fan, R.; Yi, H.; He, X.; Wang, C.; Ouyang, B.; Zhao, P.; et al. Butyrate-producing Eubacterium rectale suppresses lymphomagenesis by alleviating the TNF-induced TLR4/MyD88/NF-κB axis. Cell Host Microbe 2022, 30, 1139–1150.e7. [Google Scholar] [CrossRef]
- Vital, M.; Karch, A.; Pieper, D.H. Colonic butyrate-producing communities in humans: An overview using omics data. mSystems 2017, 2, e00130-e17. [Google Scholar] [CrossRef]
- Horwat, P.; Kopeć, S.; Garczyk, A.; Kaliciak, I.; Staręga, Z.; Drogowski, K.; Mardas, M.; Stelmach-Mardas, M. Influence of enteral nutrition on gut microbiota composition in patients with Crohn’s disease: A systematic review. Nutrients 2020, 12, 2551. [Google Scholar] [CrossRef]
- Vernocchi, P.; Del Chierico, F.; Quagliariello, A.; Ercolini, D.; Lucidi, V.; Putignani, L. A metagenomic and in Silico functional prediction of gut microbiota profiles may concur in discovering new cystic fibrosis patient-targeted probiotics. Nutrients 2017, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Dominguez Rieg, J.A.; Thomas, L.; White, J.R.; Rieg, T. Intestine-specific NHE3 deletion in adulthood causes microbial dysbiosis. Front. Cell. Infect. Microbiol. 2022, 12, 896309. [Google Scholar] [CrossRef]
- Wu, S.; Liu, Y.; Duan, Y.; Wang, F.; Guo, F.; Yan, F.; Yang, X.; Yang, X. Intestinal toxicity of deoxynivalenol is limited by supplementation with Lactobacillus plantarum JM113 and consequentially altered gut microbiota in broiler chickens. J. Anim. Sci. Biotechnol. 2018, 9, 74. [Google Scholar] [CrossRef]
- Lian, B.; He, S.; Jiang, H.; Guo, Y.; Cui, X.; Jiang, T.; Su, R.; Chen, Y.; Zhao, C.; Zhang, M.; et al. Qin-Qiao-Xiao-Du formula alleviate influenza virus infectious pneumonia through regulation gut microbiota and metabolomics. Front. Med. 2022, 9, 1032127. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, J.; Cao, Q.; Zhang, C.; Dong, Z.; Feng, D.; Ye, H.; Zuo, J. Dietary catalase supplementation alleviates deoxynivalenol-induced oxidative stress and gut microbiota dysbiosis in broiler chickens. Toxins 2022, 14, 830. [Google Scholar] [CrossRef]
- Chen, Y.; Akhtar, M.; Ma, Z.; Hu, T.; Liu, Q.; Pan, H.; Zhang, X.; Nafady, A.A.; Ansari, A.R.; Abdel-Kafy, E.-S.M.; et al. Chicken cecal microbiota reduces abdominal fat deposition by regulating fat metabolism. Npj Biofilms Microbiomes 2023, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.F., Jr.; Rôças, I.N. Pseudoramibacter alactolyticus in primary endodontic infections. J. Endod. 2003, 29, 735–738. [Google Scholar] [CrossRef]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of gut microbiota, bile acid metabolism, and cytokines in intrahepatic cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef]
- Sun, X.-W.; Abdugheni, R.; Huang, H.-J.; Wang, Y.-J.; Jiang, M.-Z.; Liu, C.; Zhou, N.; Jiang, H.; Liu, S.-J. Bacteroides propionicigenes sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2022, 72, 005397. [Google Scholar] [CrossRef]
- Huang, H.J.; Liu, C.; Sun, X.W.; Wei, R.Q.; Liu, L.W.; Chen, H.Y.; Abdugheni, R.; Wang, C.Y.; Wang, X.M.; Jiang, H.; et al. The rheumatoid arthritis gut microbial biobank reveals core microbial species that associate and effect on host inflammation and autoimmune responses. Imeta 2024, 3, e242. [Google Scholar] [CrossRef]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, H.; Liu, G.; Sha, W. First report on the bacterial diversity in the distal gut of dholes (Cuon alpinus) by using 16S rRNA gene sequences analysis. J. Appl. Genet. 2016, 57, 275–283. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Biol. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Zuo, G. CVTree: A parallel alignment-free phylogeny and taxonomy tool based on composition vectors of genomes. Genom. Proteom. Bioinform. 2021, 19, 662–667. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Auch, A.F.; von Jan, M.; Klenk, H.-P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genom. Sci. 2010, 2, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-m.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef] [PubMed]
- Pascal Andreu, V.; Augustijn, H.E.; Chen, L.; Zhernakova, A.; Fu, J.; Fischbach, M.A.; Dodd, D.; Medema, M.H. gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. 2023, 41, 1416–1423. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Allen, T.D.; Caldwell, M.E.; Lawson, P.A.; Huhnke, R.L.; Tanner, R.S. Alkalibaculum bacchi gen. nov., sp. nov., a CO-oxidizing, ethanol-producing acetogen isolated from livestock-impacted soil. Int. J. Syst. Evol. Microbiol. 2010, 60, 2483–2489. [Google Scholar] [CrossRef]
- Khomyakova, M.A.; Merkel, A.Y.; Petrova, D.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Alkalibaculum sporogenes sp. nov., isolated from a terrestrial mud volcano and emended description of the genus Alkalibaculum. Int. J. Syst. Evol. Microbiol. 2020, 70, 4914–4919. [Google Scholar] [CrossRef]
- Khomyakova, M.; Merkel, A.; Novikov, A.; Klyukina, A.; Slobodkin, A. Alkalibacter mobilis sp. nov., an anaerobic bacterium isolated from a coastal lake. Int. J. Syst. Evol. Microbiol. 2021, 71, 005174. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martínez, I.; Just, S.; Ziegler, C.; et al. The mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [PubMed]
- Genthner, B.R.S.; Bryant, M.P. Additional characteristics of one-carbon-compound utilization by Eubacterium limosum and Acetobacterium woodii. Appl. Environ. Microbiol. 1987, 53, 471–476. [Google Scholar] [CrossRef]
- Genthner, B.R.; Davis, C.L.; Bryant, M.P. Features of rumen and sewage sludge strains of Eubacterium limosum, a methanol- and H2-CO2-utilizing species. Appl. Environ. Microbiol. 1981, 42, 12–19. [Google Scholar] [CrossRef]
- Genthner, B.R.S.; Bryant, M.P. Growth of Eubacterium limosum with carbon monoxide as the energy source. Appl. Environ. Microbiol. 1982, 43, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Mountfort, D.O.; Grant, W.D.; Clarke, R.; Asher, R.A. Eubacterium callanderi sp. nov. that demethoxylates O-methoxylated aromatic acids to volatile fatty acids. Int. J. Syst. Evol. Microbiol. 1988, 38, 254–258. [Google Scholar] [CrossRef]
- Mechichi, T.; Labat, M.; Woo, T.H.S.; Thomas, P.; Garcia, J.-L.; Patel, B.K.C. Eubacterium aggreganssp. nov., a new homoacetogenic bacterium from olive mill wastewater treatment digestor. Anaerobe 1998, 4, 283–291. [Google Scholar] [CrossRef]
- Collins, M.D.; Lawson, P.A.; Willems, A.; Cordoba, J.J.; Fernandez-Garayzabal, J.; Garcia, P.; Cai, J.; Hippe, H.; Farrow, J.A. The phylogeny of the genus Clostridium: Proposal of five new genera and eleven new species combinations. Int. J. Syst. Evol. Microbiol. 1994, 44, 812–826. [Google Scholar] [CrossRef]
- Fujita, Y.; Matsuoka, H.; Hirooka, K. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 2007, 66, 829–839. [Google Scholar] [CrossRef]
- Tan, Z.; Black, W.; Yoon, J.M.; Shanks, J.V.; Jarboe, L.R. Improving Escherichia coli membrane integrity and fatty acid production by expression tuning of FadL and OmpF. Microb. Cell Factories 2017, 16, 38. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.-L.; Xie, B.-B.; Zhang, X.-Y.; Chen, X.-L.; Zhou, B.-C.; Zhou, J.; Oren, A.; Zhang, Y.-Z. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Sasi Jyothsna, T.S.; Tushar, L.; Sasikala, C.; Ramana, C.V. Paraclostridium benzoelyticum gen. nov., sp. nov., isolated from marine sediment and reclassification of Clostridium bifermentans as Paraclostridium bifermentans comb. nov. Proposal of a new genus Paeniclostridium gen. nov. to accommodate Clostridium sordellii and Clostridium ghonii. Int. J. Syst. Evol. Microbiol. 2016, 66, 1268–1274. [Google Scholar]
- Christensen, S.J.; Madsen, M.S.; Zinck, S.S.; Hedberg, C.; Sørensen, O.B.; Svensson, B.; Meyer, A.S. Enzymatic potato starch modification and structure-function analysis of six diverse GH77 4-alpha-glucanotransferases. Int. J. Biol. Macromol. 2023, 224, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Henrissat, B.; Gouret, P.; Pontarotti, P.; Drancourt, M. Peptidoglycan: A post-genomic analysis. BMC Microbiol. 2012, 12, 294. [Google Scholar] [CrossRef]
- Ulaganathan, T.; Shi, R.; Yao, D.; Gu, R.-X.; Garron, M.-L.; Cherney, M.; Tieleman, D.P.; Sterner, E.; Li, G.; Li, L.; et al. Conformational flexibility of PL12 family heparinases: Structure and substrate specificity of heparinase III from Bacteroides thetaiotaomicron (BT4657). Glycobiology 2017, 27, 176–187. [Google Scholar] [CrossRef]
- Badieyan, S.; Bevan, D.R.; Zhang, C. Study and design of stability in GH5 cellulases. Biotechnol. Bioeng. 2012, 109, 31–44. [Google Scholar] [CrossRef]
- Nath, P.; Goyal, A. Structure and dynamics analysis of multi-domain putative β-1,4-glucosidase of family 3 glycoside hydrolase (PsGH3) from Pseudopedobacter saltans. J. Mol. Model. 2021, 27, 106. [Google Scholar] [CrossRef]
- Gavande, P.V.; Goyal, A. Chapter 6—Endo-β-1,3-glucanase. In Glycoside Hydrolases; Goyal, A., Sharma, K., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 121–133. [Google Scholar]
- Blachier, F.; Mariotti, F.; Huneau, J.F.; Tomé, D. Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids 2007, 33, 547–562. [Google Scholar] [CrossRef]
- Luo, W.; Zhao, M.; Dwidar, M.; Gao, Y.; Xiang, L.; Wu, X.; Medema, M.H.; Xu, S.; Li, X.; Schäfer, H.; et al. Microbial assimilatory sulfate reduction-mediated H2S: An overlooked role in Crohn’s disease development. Microbiome 2024, 12, 152. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Martín, R.; Rossi, O.; Bermúdez-Humarán, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ouyang, L.; Wang, W.; Chen, B.; Liu, W.; Yuan, X.; Luo, Y.; Cheng, T.; Yeung, K.W.K.; Liu, X.; et al. Sodium butyrate-modified sulfonated polyetheretherketone modulates macrophage behavior and shows enhanced antibacterial and osteogenic functions during implant-associated infections. J. Mater. Chem. B 2019, 7, 5541–5553. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jin, D.; Huang, S.; Wu, J.; Xu, M.; Liu, T.; Dong, W.; Liu, X.; Wang, S.; Zhong, W.; et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020, 469, 456–467. [Google Scholar] [CrossRef]
- Wang, R.; Yang, X.; Liu, J.; Zhong, F.; Zhang, C.; Chen, Y.; Sun, T.; Ji, C.; Ma, D. Gut microbiota regulates acute myeloid leukaemia via alteration of intestinal barrier function mediated by butyrate. Nat. Commun. 2022, 13, 2522. [Google Scholar] [CrossRef]
- Lau, H.C.-H.; Zhang, X.; Ji, F.; Lin, Y.; Liang, W.; Li, Q.; Chen, D.; Fong, W.; Kang, X.; Liu, W.; et al. Lactobacillus acidophilus suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma through producing valeric acid. eBioMedicine 2024, 100, 104952. [Google Scholar] [CrossRef]
- Bindu, K.; Usha, K.; Dhananjay Kumar, S.; Gulam Mohammed, H.; Dinesh Kumar, P.; Anshul, S.; Ravi Bhushan, S.; Gyan Prakash, M.; Gireesh Kumar, S. Molecular targets of valeric acid: A bioactive natural product for endocrine, metabolic, and immunological disorders. Endocr. Metab. Immune Disord. Drug Targets 2024, 24, 1506–1517. [Google Scholar]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [PubMed]
- Duan, H.; Wang, L.; Huangfu, M.; Li, H. The impact of microbiota-derived short-chain fatty acids on macrophage activities in disease: Mechanisms and therapeutic potentials. Biomed. Pharmacother. 2023, 165, 115276. [Google Scholar]
- Choi, M.H.; Kim, D.; Lee, K.H.; Kim, H.J.; Sul, W.J.; Jeong, S.H. Dysbiosis of the gut microbiota is associated with in-hospital mortality in patients with antibiotic-associated diarrhoea: A metagenomic analysis. Int. J. Antimicrob. Agents 2024, 64, 107330. [Google Scholar] [CrossRef]
- Masoodi, I.; Ahmad, S.; Alyamani, E.J.; Al-Lehibi, A.A.; Qutub, A.N.; Alsayari, K.N.; Alomair, A.O. Microbial dysbiosis in inflammatory bowel diseases: Results of a metagenomic study in Saudi Arabia. Minerva Gastroenterol. Dietol. 2019, 65, 177–186. [Google Scholar] [CrossRef]
- Liao, Y.; Wu, F.; Dai, F.; Huang, Q.; Feng, Y.; Ling, Y.; Lu, H. Mycobacterium tuberculosis and Pseudoramibacter alactolyticus coinfection in brain after dental extraction: A case report. Medicine 2019, 98, e18289. [Google Scholar] [PubMed]
- Liang, J.; Zhao, Y.; Xi, Y.; Xiang, C.; Yong, C.; Huo, J.; Zou, H.; Hou, Y.; Pan, Y.; Wu, M.; et al. Association between depression, anxiety symptoms and gut microbiota in Chinese elderly with functional constipation. Nutrients 2022, 14, 5013. [Google Scholar] [CrossRef]
- Rimal, B.; Collins, S.L.; Tanes, C.E.; Rocha, E.R.; Granda, M.A.; Solanki, S.; Hoque, N.J.; Gentry, E.C.; Koo, I.; Reilly, E.R.; et al. Bile salt hydrolase catalyses formation of amine-conjugated bile acids. Nature 2024, 626, 859–863. [Google Scholar] [CrossRef]
- Stummer, N.; Feichtinger, R.G.; Weghuber, D.; Kofler, B.; Schneider, A.M. Role of hydrogen sulfide in inflammatory bowel disease. Antioxidants 2023, 12, 1570. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Millan, M.J.; Leite, G.; Wang, J.; Morales, W.; Parodi, G.; Pimentel, M.L.; Barlow, G.M.; Mathur, R.; Rezaie, A.; Sanchez, M.; et al. Methanogens and hydrogen sulfide producing bacteria guide distinct gut microbe profiles and irritable bowel syndrome subtypes. Off. J. Am. Coll. Gastroenterol. ACG 2022, 117, 2055–2066. [Google Scholar] [CrossRef]
- Fang, Y.; Yan, C.; Zhao, Q.; Xu, J.; Liu, Z.; Gao, J.; Zhu, H.; Dai, Z.; Wang, D.; Tang, D. The roles of microbial products in the development of colorectal cancer: A review. Bioengineered 2021, 12, 720–735. [Google Scholar]
- Teigen, L.M.; Geng, Z.; Sadowsky, M.J.; Vaughn, B.P.; Hamilton, M.J.; Khoruts, A. Dietary factors in sulfur metabolism and pathogenesis of ulcerative colitis. Nutrients 2019, 11, 931. [Google Scholar] [CrossRef]
- Wu, D.; Luo, N.; Wang, L.; Zhao, Z.; Bu, H.; Xu, G.; Yan, Y.; Che, X.; Jiao, Z.; Zhao, T.; et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways. Sci. Rep. 2017, 7, 455. [Google Scholar] [CrossRef] [PubMed]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid beta-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Dordević, D.; Vítězová, M. Possible synergy effect of hydrogen sulfide and acetate produced by sulfate-reducing bacteria on inflammatory bowel disease development. J. Adv. Res. 2021, 27, 71–78. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, R.; Zhou, X.; Ji, F.; Zhang, B. Hydrogen sulfide improves colonic barrier integrity in DSS-induced inflammation in Caco-2 cells and mice. Int. Immunopharmacol. 2016, 39, 121–127. [Google Scholar] [PubMed]
- Lin, M.; Hu, G.; Yu, B. Dysregulated cystathionine-β-synthase/hydrogen sulfide signaling promotes chronic stress-induced colonic hypermotility in rats. Neurogastroenterol. Motil. 2023, 35, e14488. [Google Scholar] [CrossRef]
- Zhang, W.; An, Y.; Qin, X.; Wu, X.; Wang, X.; Hou, H.; Song, X.; Liu, T.; Wang, B.; Huang, X.; et al. Gut microbiota-derived metabolites in colorectal cancer: The bad and the challenges. Front. Oncol. 2021, 11, 739648. [Google Scholar] [CrossRef]
- Gao, H.-H.; Zhao, S.; Wang, R.-J.; Qin, D.-Y.; Chen, P.; Zhang, A.-S.; Zhuang, Q.-Y.; Zhai, Y.-F.; Zhou, X.-H. Gut bacterium promotes host fitness in special ecological niche by affecting sugar metabolism in Drosophila suzukii. Insect Sci. 2023, 30, 1713–1733. [Google Scholar]
- Hudry, B.; de Goeij, E.; Mineo, A.; Gaspar, P.; Hadjieconomou, D.; Studd, C.; Mokochinski, J.B.; Kramer, H.B.; Plaçais, P.-Y.; Preat, T.; et al. Sex differences in intestinal carbohydrate metabolism promote food intake and sperm maturation. Cell 2019, 178, 901–918.e16. [Google Scholar] [CrossRef]
- Hicks, K.G.; Cluntun, A.A.; Schubert, H.L.; Hackett, S.R.; Berg, J.A.; Leonard, P.G.; Ajalla Aleixo, M.A.; Zhou, Y.; Bott, A.J.; Salvatore, S.R.; et al. Protein-metabolite interactomics of carbohydrate metabolism reveal regulation of lactate dehydrogenase. Science 2023, 379, 996–1003. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolation sources | Human feces | Human feces | Livestock-impacted soil | Terrestrial mud volcano | Coastal lake | Muris of a mouse | Human feces | Dental calculus | Aachen minipig | Human feces | Anaerobic digestor | Human feces |
| Motility | − | ND | + | + | + | ND | − | − | ND | − | − | − |
| Aesculin/Esculin hydrolysis | + | − | − | ND | ND | + | + | − | ND | + | − | + |
| Acid production from: | ||||||||||||
| Glucose | + | W | + | + | + | − | − | + | ND | + | + | + |
| Xylose | + | W | − | + | − | − | − | − | ND | W | − | − |
| Glycerol | − | ND | − | − | − | − | − | − | ND | − | + | + |
| Cellobiose | − | ND | − | − | − | − | − | − | ND | − | − | − |
| Maltose | − | − | − | − | − | − | − | − | ND | − | − | − |
| Mannitol | − | − | − | ND | ND | − | + | + | ND | + | ND | + |
| Mannose | − | − | + | + | + | − | − | − | ND | − | − | − |
| Sucrose | − | − | − | − | + | − | − | − | ND | − | ND | − |
| Lactose | − | − | − | − | − | − | − | − | ND | − | − | − |
| Enzyme activities: | ||||||||||||
| Esterase C4 | + | W | W | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Esterase lipase C8 | + | W | − | ND | ND | ND | + | ND | ND | ND | ND | ND |
| Trypsin | − | − | − | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Catalase | − | − | ND | − | ND | ND | − | ND | ND | − | ND | ND |
| β-Glucosidase | − | − | − | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Alkaline phosphatase | + | − | − | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Naphthol-AS-Bi-phosphohydrolase | + | W | W | ND | ND | ND | + | ND | ND | ND | ND | ND |
| N-Acetyl-β-glucosaminidase | − | − | W | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Valine arylamidase | W | − | − | ND | ND | ND | − | ND | ND | ND | ND | ND |
| Cystine arylamidase | W | − | − | ND | ND | ND | − | ND | ND | ND | ND | ND |
| DNA G + C content (mol%) | 52.0 mol% | 70.0 mol% | 34.0 mol% | 32.3 mol% | 39.1 mol% | 35.8 mol% | 29.8 mol% | 61.0 mol% | 49.3 mol% | 47.2 mol% | 47.0 mol% | 45.0 mol% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.-M.; Huang, H.-J.; Sun, X.-W.; Wei, R.-Q.; Chen, H.-Y.; Liu, C.; Liu, S.-J. Identification and Characterization of Two Novel Members of the Family Eubacteriaceae, Anaerofustis butyriciformans sp. nov. and Pseudoramibacter faecis sp. nov., Isolated from Human Feces. Microorganisms 2025, 13, 916. https://doi.org/10.3390/microorganisms13040916
Wang X-M, Huang H-J, Sun X-W, Wei R-Q, Chen H-Y, Liu C, Liu S-J. Identification and Characterization of Two Novel Members of the Family Eubacteriaceae, Anaerofustis butyriciformans sp. nov. and Pseudoramibacter faecis sp. nov., Isolated from Human Feces. Microorganisms. 2025; 13(4):916. https://doi.org/10.3390/microorganisms13040916
Chicago/Turabian StyleWang, Xiao-Meng, Hao-Jie Huang, Xin-Wei Sun, Rui-Qi Wei, Hao-Yu Chen, Chang Liu, and Shuang-Jiang Liu. 2025. "Identification and Characterization of Two Novel Members of the Family Eubacteriaceae, Anaerofustis butyriciformans sp. nov. and Pseudoramibacter faecis sp. nov., Isolated from Human Feces" Microorganisms 13, no. 4: 916. https://doi.org/10.3390/microorganisms13040916
APA StyleWang, X.-M., Huang, H.-J., Sun, X.-W., Wei, R.-Q., Chen, H.-Y., Liu, C., & Liu, S.-J. (2025). Identification and Characterization of Two Novel Members of the Family Eubacteriaceae, Anaerofustis butyriciformans sp. nov. and Pseudoramibacter faecis sp. nov., Isolated from Human Feces. Microorganisms, 13(4), 916. https://doi.org/10.3390/microorganisms13040916