
Microbiome Migration from Soil to Leaves in Maize and Rice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Seed Germination, and Growth Conditions
2.2. Sample Collection
2.3. DNA Extraction, PCR Amplification, and Sequencing
2.4. Bioinformatics Analysis
2.5. Isolation of Bacteria
2.6. Synthetic Community Assembly and Inoculation
3. Results
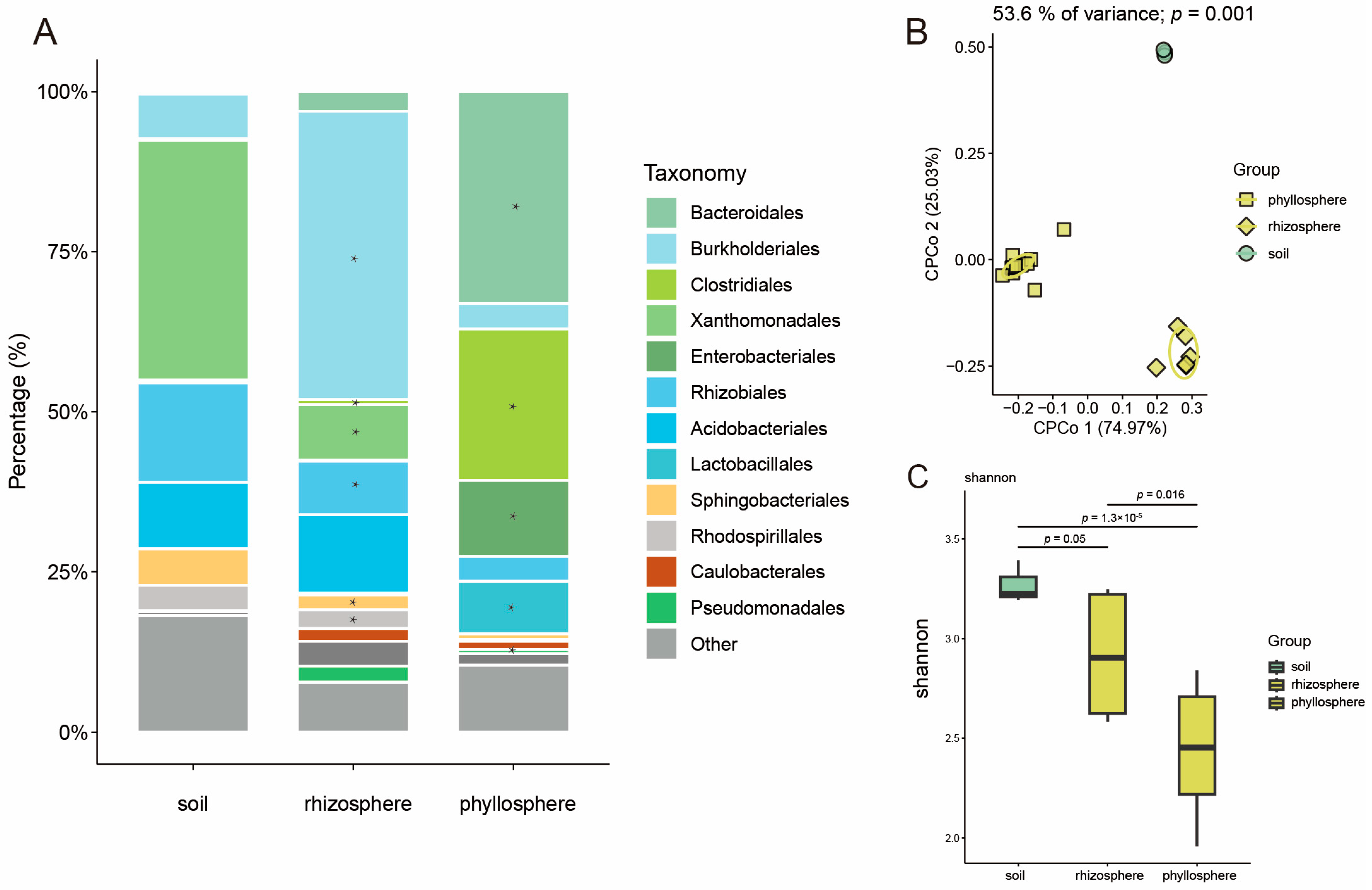
3.1. The Pattern of Microbiome Migration Varies Between Rice and Maize
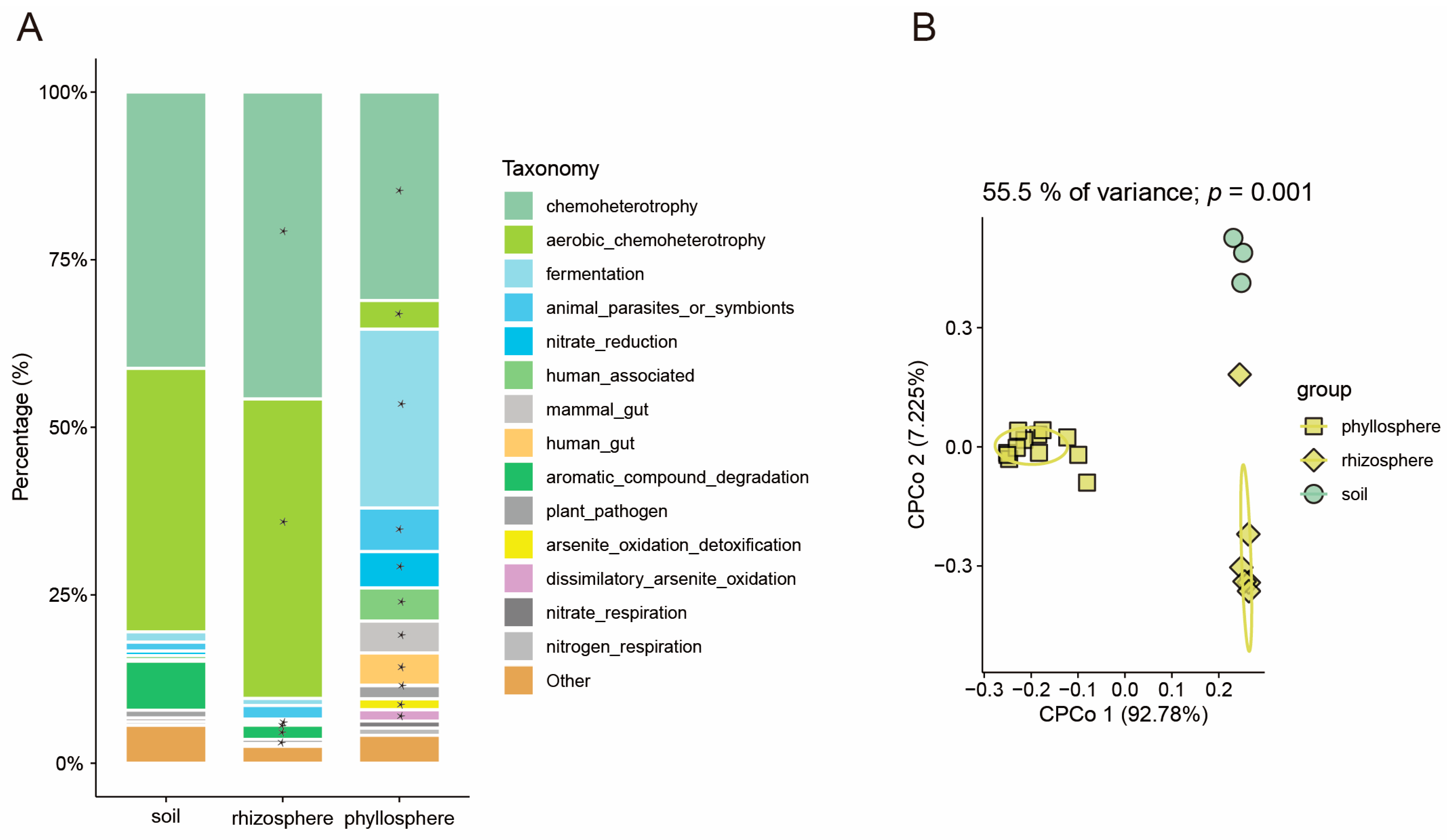
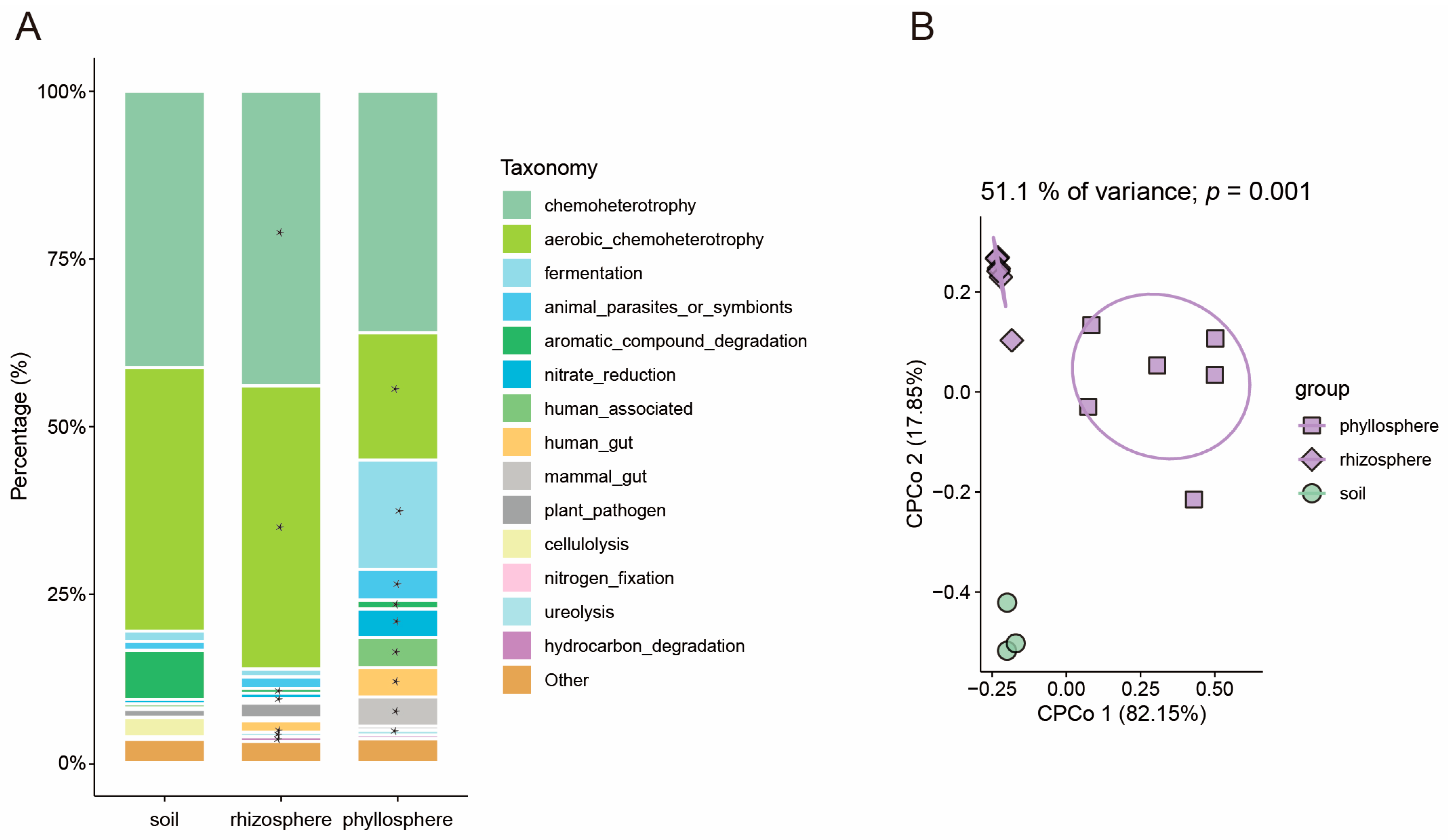
3.2. The Pattern of Microbiome Function Migration Varies Between Rice and Maize
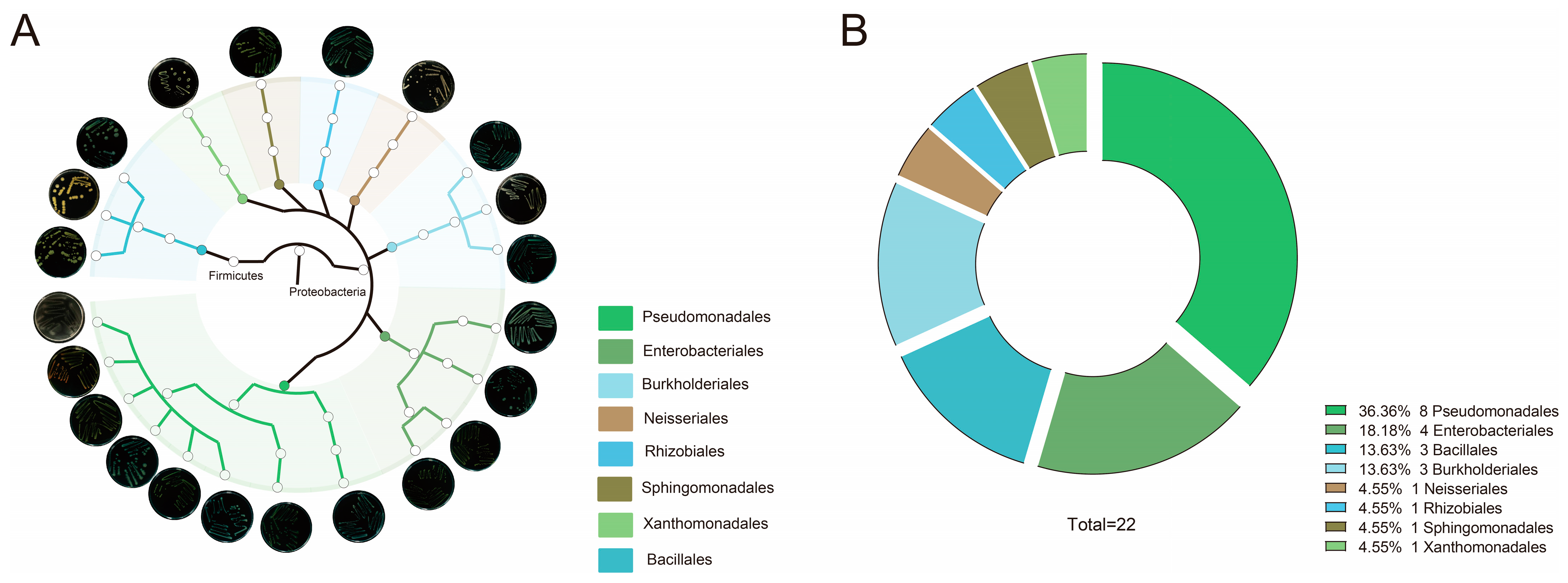
3.3. Isolation of Microbiota from the Phyllosphere After Migration Events
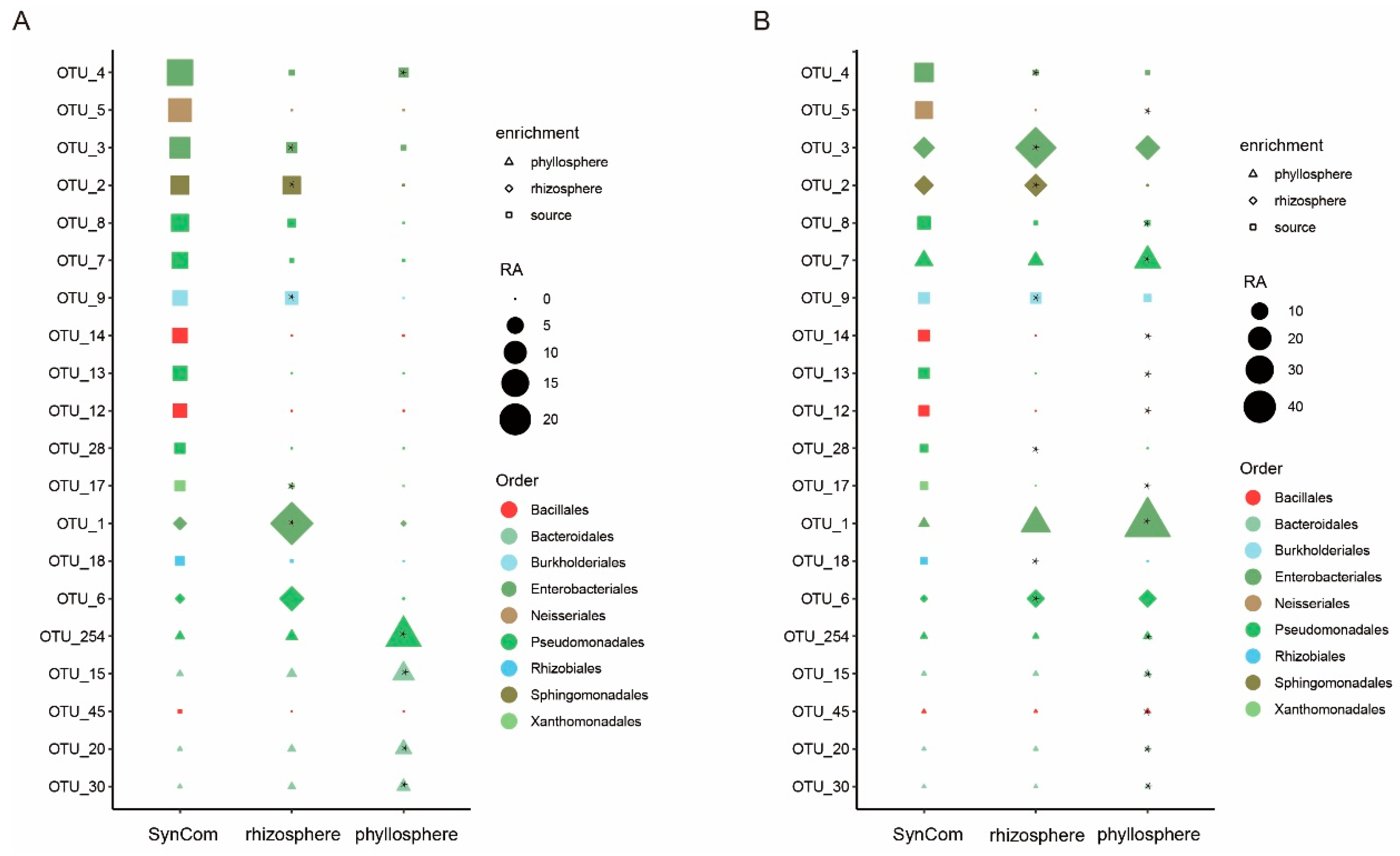
3.4. Investigatione of SynCom Migration to Rhizosphere and Phyllosphere of Rice and Maize
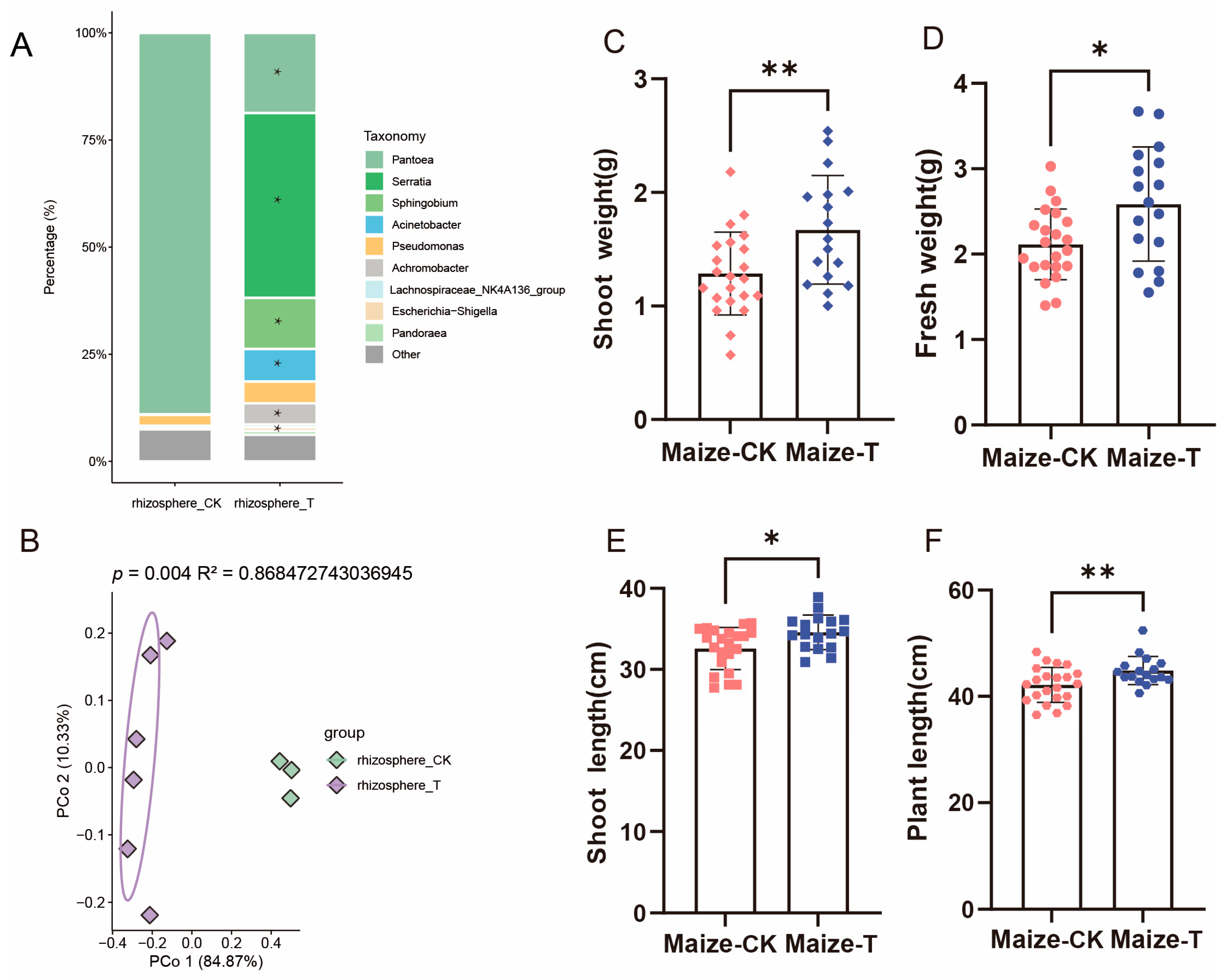
3.5. Rice and Maize Can Assemble Microbiota from SynCom to Promote Plant Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Getzke, F.; Thiergart, T.; Hacquard, S. Contribution of bacterial-fungal balance to plant and animal health. Curr. Opin. Microbiol. 2019, 49, 66–72. [Google Scholar] [CrossRef]
- Compant, S.; Cambon, M.C.; Vacher, C.; Mitter, B.; Samad, A.; Sessitsch, A. The plant endosphere world—Bacterial life within plants. Environ. Microbiol. 2021, 23, 1812–1829. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mazahar, S.; Chapadgaonkar, S.S.; Giri, P.; Shourie, A. Phyto-microbiome to mitigate abiotic stress in crop plants. Front. Microbiol. 2023, 14, 1210890. [Google Scholar] [CrossRef]
- Zhang, J.; Cook, J.; Nearing, J.T.; Zhang, J.; Raudonis, R.; Glick, B.R.; Langille, M.G.I.; Cheng, Z. Harnessing the plant microbiome to promote the growth of agricultural crops. Microbiol. Res. 2021, 245, 126690. [Google Scholar] [CrossRef]
- Pal, G.; Saxena, S.; Kumar, K.; Verma, A.; Sahu, P.K.; Pandey, A.; White, J.F.; Verma, S.K. Endophytic Burkholderia: Multifunctional roles in plant growth promotion and stress tolerance. Microbiol. Res. 2022, 265, 127201. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-San Millan, A.; Farran, I.; Larraya, L.; Ancin, M.; Arregui, L.M.; Veramendi, J. Plant growth-promoting traits of yeasts isolated from Spanish vineyards: Benefits for seedling development. Microbiol. Res. 2020, 237, 126480. [Google Scholar] [CrossRef] [PubMed]
- Bettenfeld, P.; Cadena, I.C.J.; Jacquens, L.; Fernandez, O.; Fontaine, F.; van Schaik, E.; Courty, P.E.; Trouvelot, S. The microbiota of the grapevine holobiont: A key component of plant health. J. Adv. Res. 2022, 40, 1–15. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, Z.; Liu, J.; Li, X.; Wang, X.; Dai, C.; Zhang, T.; Carrión, V.J.; Wei, Z.; Cao, F.; et al. Crop rotation and native microbiome inoculation restore soil capacity to suppress a root disease. Nat. Commun. 2023, 14, 8126. [Google Scholar] [CrossRef]
- Van Deynze, A.; Zamora, P.; Delaux, P.M.; Heitmann, C.; Jayaraman, D.; Rajasekar, S.; Graham, D.; Maeda, J.; Gibson, D.; Schwartz, K.D.; et al. Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota. PLoS Biol. 2018, 16, e2006352. [Google Scholar] [CrossRef]
- Pang, F.; Li, Q.; Solanki, M.K.; Wang, Z.; Xing, Y.X.; Dong, D.F. Soil phosphorus transformation and plant uptake driven by phosphate-solubilizing microorganisms. Front. Microbiol. 2024, 15, 1383813. [Google Scholar] [CrossRef]
- Vurukonda, S.S.; Vardharajula, S.; Shrivastava, M.; Sk, Z.A. Enhancement of drought stress tolerance in crops by plant growth promoting rhizobacteria. Microbiol. Res. 2016, 184, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Malacrinò, A.; Mosca, S.; Li Destri Nicosia, M.G.; Agosteo, G.E.; Schena, L. Plant Genotype Shapes the Bacterial Microbiome of Fruits, Leaves, and Soil in Olive Plants. Plants 2022, 11, 613. [Google Scholar] [CrossRef]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Compant, S.; van der Heijden, M.G.; Sessitsch, A. Climate change effects on beneficial plant-microorganism interactions. FEMS Microbiol. Ecol. 2010, 73, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Morales Moreira, Z.P.; Helgason, B.L.; Germida, J.J. Crop, genotype, and field environmental conditions shape bacterial and fungal seed epiphytic microbiomes. Can. J. Microbiol. 2021, 67, 161–173. [Google Scholar] [CrossRef]
- Xie, Y.; Ouyang, Y.; Han, S.; Se, J.; Tang, S.; Yang, Y.; Ma, Q.; Wu, L. Crop rotation stage has a greater effect than fertilisation on soil microbiome assembly and enzymatic stoichiometry. Sci. Total Environ. 2022, 815, 152956. [Google Scholar] [CrossRef]
- Gong, T.; Xin, X.F. Phyllosphere microbiota: Community dynamics and its interaction with plant hosts. J. Integr. Plant Biol. 2021, 63, 297–304. [Google Scholar] [CrossRef]
- Korenblum, E.; Massalha, H.; Aharoni, A. Plant-microbe interactions in the rhizosphere via a circular metabolic economy. Plant Cell 2022, 34, 3168–3182. [Google Scholar] [CrossRef]
- Tkacz, A.; Bestion, E.; Bo, Z.; Hortala, M.; Poole, P.S. Influence of Plant Fraction, Soil, and Plant Species on Microbiota: A Multikingdom Comparison. mBio 2020, 11, e02785-19. [Google Scholar] [CrossRef]
- Wei, N.; Ashman, T.L. The effects of host species and sexual dimorphism differ among root, leaf and flower microbiomes of wild strawberries in situ. Sci. Rep. 2018, 8, 5195. [Google Scholar] [CrossRef]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef]
- Park, I.; Seo, Y.S.; Mannaa, M. Recruitment of the rhizo-microbiome army: Assembly determinants and engineering of the rhizosphere microbiome as a key to unlocking plant potential. Front. Microbiol. 2023, 14, 1163832. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xu, Z.; Liu, H.; Liu, Y.; Zhou, Y.; Meng, C.; Ma, S.; Xie, Z.; Li, Y.; Zhang, C.S. Patterns in the Microbial Community of Salt-Tolerant Plants and the Functional Genes Associated with Salt Stress Alleviation. Microbiol. Spectr. 2021, 9, e00767-21. [Google Scholar] [CrossRef] [PubMed]
- Vives-Peris, V.; de Ollas, C.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root exudates: From plant to rhizosphere and beyond. Plant Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Stringlis, I.A.; van Bentum, S.; de Jonge, R.; Snoek, B.L.; Pieterse, C.M.J.; Bakker, P.; Berendsen, R.L. Transcriptome Signatures in Pseudomonas simiae WCS417 Shed Light on Role of Root-Secreted Coumarins in Arabidopsis-Mutualist Communication. Microorganisms 2021, 9, 575. [Google Scholar] [CrossRef]
- Massoni, J.; Bortfeld-Miller, M.; Widmer, A.; Vorholt, J.A. Capacity of soil bacteria to reach the phyllosphere and convergence of floral communities despite soil microbiota variation. Proc. Natl. Acad. Sci. USA 2021, 118, e2100150118. [Google Scholar] [CrossRef]
- Frindte, K.; Zoche, S.A.; Knief, C. Development of a Distinct Microbial Community Upon First Season Crop Change in Soils of Long-Term Managed Maize and Rice Fields. Front. Microbiol. 2020, 11, 588198. [Google Scholar] [CrossRef]
- Wossen, T.; Menkir, A.; Alene, A.; Abdoulaye, T.; Ajala, S.; Badu-Apraku, B.; Gedil, M.; Mengesha, W.; Meseka, S. Drivers of transformation of the maize sector in Nigeria. Glob. Food Secur. 2023, 38, 100713. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, Y.; Shi, H.; Qiu, J.; Ding, X.; Kou, Y. Recent Progress in Rice Broad-Spectrum Disease Resistance. Int. J. Mol. Sci. 2021, 22, 11658. [Google Scholar] [CrossRef]
- Sanna, M.; Martino, I.; Guarnaccia, V.; Mezzalama, M. Diversity and Pathogenicity of Fusarium Species Associated with Stalk and Crown Rot in Maize in Northern Italy. Plants 2023, 12, 3857. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Prakash, A.; Johri, B.N. Induced systemic resistance (ISR) in plants: Mechanism of action. Indian J. Microbiol. 2007, 47, 289–297. [Google Scholar] [CrossRef]
- Hata, E.M.; Yusof, M.T.; Zulperi, D. Induction of Systemic Resistance against Bacterial Leaf Streak Disease and Growth Promotion in Rice Plant by Streptomyces shenzhenesis TKSC3 and Streptomyces sp. SS8. Plant Pathol. J. 2021, 37, 173–181. [Google Scholar] [CrossRef]
- Saravanakumar, K.; Li, Y.; Yu, C.; Wang, Q.Q.; Wang, M.; Sun, J.; Gao, J.X.; Chen, J. Effect of Trichoderma harzianum on maize rhizosphere microbiome and biocontrol of Fusarium Stalk rot. Sci. Rep. 2017, 7, 1771. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Q.; Liu, J.; Wang, L.; Wu, X.; Zhao, Z.; Wang, N.; Gao, Z. Suaeda salsa Root-Associated Microorganisms Could Effectively Improve Maize Growth and Resistance under Salt Stress. Microbiol. Spectr. 2022, 10, e01349-22. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant-microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.; Ahmed, T.; Ijaz, U.; Shahid, M.; Azizullah; Li, D.; Manzoor, I.; Song, F. Plant-Microbiome Crosstalk: Dawning from Composition and Assembly of Microbial Community to Improvement of Disease Resilience in Plants. Int. J. Mol. Sci. 2021, 22, 6852. [Google Scholar] [CrossRef]
- Qu, Q.; Zhang, Z.; Peijnenburg, W.; Liu, W.; Lu, T.; Hu, B.; Chen, J.; Chen, J.; Lin, Z.; Qian, H. Rhizosphere Microbiome Assembly and Its Impact on Plant Growth. J. Agric. Food Chem. 2020, 68, 5024–5038. [Google Scholar] [CrossRef]
- Cadot, S.; Guan, H.; Bigalke, M.; Walser, J.C.; Jander, G.; Erb, M.; van der Heijden, M.G.A.; Schlaeppi, K. Specific and conserved patterns of microbiota-structuring by maize benzoxazinoids in the field. Microbiome 2021, 9, 103. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, L.; Wen, Y.; Zhang, M.; Huang, S.; Wang, S.; Zhao, Y.; Hao, X.; Li, L.; Gao, Q.; et al. Maize functional requirements drive the selection of rhizobacteria under long-term fertilization practices. New Phytol. 2024, 242, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Liu, Y.; Geng, J.; Lü, H.; Zhao, H.M.; Xiang, L.; Li, H.; Mo, C.H.; Li, Y.W.; Cai, Q.Y. Maize root-associated niches determine the response variation in bacterial community assembly and function to phthalate pollution. J. Hazard. Mater. 2022, 429, 128280. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Dai, R.; Xu, H.; Liu, Y.; Bai, B.; Meng, Y.; Li, H.; Cao, X.; Bai, Y.; Song, X.; et al. The rice histone methylation regulates hub species of the root microbiota. J. Genet. Genom. 2021, 48, 836–843. [Google Scholar] [CrossRef]
- Wang, X.; He, S.W.; He, Q.; Ju, Z.C.; Ma, Y.N.; Wang, Z.; Han, J.C.; Zhang, X.X. Early inoculation of an endophyte alters the assembly of bacterial communities across rice plant growth stages. Microbiol. Spectr. 2023, 11, e04978-22. [Google Scholar] [CrossRef] [PubMed]
- Grosskopf, T.; Soyer, O.S. Synthetic microbial communities. Curr. Opin. Microbiol. 2014, 18, 72–77. [Google Scholar] [CrossRef]
- Alzate Zuluaga, M.Y.; Fattorini, R.; Cesco, S.; Pii, Y. Plant-microbe interactions in the rhizosphere for smarter and more sustainable crop fertilization: The case of PGPR-based biofertilizers. Front. Microbiol. 2024, 15, 1440978. [Google Scholar] [CrossRef]
- Xu, X.; Dinesen, C.; Pioppi, A.; Kovács, Á.T.; Lozano-Andrade, C.N. Composing a microbial symphony: Synthetic communities for promoting plant growth. Trends Microbiol. 2025, S0966-842X(25)00006-X. [Google Scholar] [CrossRef]
- Schmitz, L.; Yan, Z.; Schneijderberg, M.; de Roij, M.; Pijnenburg, R.; Zheng, Q.; Franken, C.; Dechesne, A.; Trindade, L.M.; van Velzen, R.; et al. Synthetic bacterial community derived from a desert rhizosphere confers salt stress resilience to tomato in the presence of a soil microbiome. ISME J. 2022, 16, 1907–1920. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Huang, J.; Zhu, L.; Lu, X.; Cui, F.; Wang, J.; Zhou, C. A simplified synthetic rhizosphere bacterial community steers plant oxylipin pathways for preventing foliar phytopathogens. Plant Physiol. Biochem. 2023, 202, 107941. [Google Scholar] [CrossRef]
- De la Vega-Camarillo, E.; Sotelo-Aguilar, J.; Rios-Galicia, B.; Mercado-Flores, Y.; Arteaga-Garibay, R.; Villa-Tanaca, L.; Hernández-Rodríguez, C. Promotion of the growth and yield of Zea mays by synthetic microbial communities from Jala maize. Front. Microbiol. 2023, 14, 1167839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.; Liu, F.; Liang, J.; Zhao, P.; Tsui, C.K.M.; Cai, L. Cross-kingdom synthetic microbiota supports tomato suppression of Fusarium wilt disease. Nat. Commun. 2022, 13, 7890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, X.; Solanki, M.K.; Pang, F. A Synthetic Microbial Community of Plant Core Microbiome Can Be a Potential Biocontrol Tool. J. Agric. Food Chem. 2023, 71, 5030–5041. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Kang, H.; Peng, Q.; Wicaksono, W.A.; Berg, G.; Liu, Z.; Ma, J.; Zhang, D.; Cernava, T.; Liu, Y. Microbiome homeostasis on rice leaves is regulated by a precursor molecule of lignin biosynthesis. Nat. Commun. 2024, 15, 23. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Liu, Y.X.; Chen, L.; Ma, T.; Li, X.; Zheng, M.; Zhou, X.; Chen, L.; Qian, X.; Xi, J.; Lu, H.; et al. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2023, 2, e83. [Google Scholar] [CrossRef]
- Pfeilmeier, S.; Petti, G.C.; Bortfeld-Miller, M.; Daniel, B.; Field, C.M.; Sunagawa, S.; Vorholt, J.A. The plant NADPH oxidase RBOHD is required for microbiota homeostasis in leaves. Nat. Microbiol. 2021, 6, 852–864. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef]
- Yashiro, E.; Spear, R.N.; McManus, P.S. Culture-dependent and culture-independent assessment of bacteria in the apple phyllosphere. J. Appl. Microbiol. 2011, 110, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Li, X.; Zhang, Z.; Wang, G.; Zhang, Y.; Chen, L. Exploring microbial diversity and function in companion planting systems of white clover and orchard grass. Sci. Rep. 2024, 14, 21609. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; van der Heijden, M.G.A. Soil microbiomes and one health. Nat. Rev. Microbiol. 2023, 21, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Wistrom, C.; Lindow, S.E. A cell-cell signaling sensor is required for virulence and insect transmission of Xylella fastidiosa. Proc. Natl. Acad. Sci. USA 2008, 105, 2670–2675. [Google Scholar] [CrossRef]
- Navarro-Noya, Y.E.; Chávez-Romero, Y.; Hereira-Pacheco, S.; de León Lorenzana, A.S.; Govaerts, B.; Verhulst, N.; Dendooven, L. Bacterial Communities in the Rhizosphere at Different Growth Stages of Maize Cultivated in Soil Under Conventional and Conservation Agricultural Practices. Microbiol. Spectr. 2022, 10, e01834-21. [Google Scholar] [CrossRef]
- Murphy, K.M.; Edwards, J.; Louie, K.B.; Bowen, B.P.; Sundaresan, V.; Northen, T.R.; Zerbe, P. Bioactive diterpenoids impact the composition of the root-associated microbiome in maize (Zea mays). Sci. Rep. 2021, 11, 333. [Google Scholar] [CrossRef]
- Chang, J.; Tian, L.; Leite, M.F.A.; Sun, Y.; Shi, S.; Xu, S.; Wang, J.; Chen, H.; Chen, D.; Zhang, J.; et al. Nitrogen, manganese, iron, and carbon resource acquisition are potential functions of the wild rice Oryza rufipogon core rhizomicrobiome. Microbiome 2022, 10, 196. [Google Scholar] [CrossRef]
- de Albuquerque, T.M.; Mendes, L.W.; Rocha, S.M.B.; Antunes, J.E.L.; Oliveira, L.M.S.; Melo, V.M.M.; Oliveira, F.A.S.; Pereira, A.P.A.; da Silva, V.B.; Gomes, R.L.F.; et al. Genetically related genotypes of cowpea present similar bacterial community in the rhizosphere. Sci. Rep. 2022, 12, 3472. [Google Scholar] [CrossRef]
- Liu, B.; Dai, Y.; Cheng, X.; He, X.; Bei, Q.; Wang, Y.; Zhou, Y.; Zhu, B.; Zhang, K.; Tian, X.; et al. Straw mulch improves soil carbon and nitrogen cycle by mediating microbial community structure and function in the maize field. Front. Microbiol. 2023, 14, 1217966. [Google Scholar] [CrossRef]
- Zhou, Y.; He, Z.; Lin, Q.; Lin, Y.; Long, K.; Xie, Z.; Hu, W. Salt stress affects the bacterial communities in rhizosphere soil of rice. Front. Microbiol. 2024, 15, 1505368. [Google Scholar] [CrossRef]
- Liu, W.; Qiu, K.; Xie, Y.; Wang, R.; Li, H.; Meng, W.; Yang, Y.; Huang, Y.; Li, Y.; He, Y. Years of sand fixation with Caragana korshinskii drive the enrichment of its rhizosphere functional microbes by accumulating soil N. PeerJ 2022, 10, e14271. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Ran, Q.; Li, X.; Dong, C.; Huang, J.; Han, Y. Deciphering the effect of phytohormones on the phyllosphere microbiota of Eucommia ulmoides. Microbiol. Res. 2024, 278, 127513. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wang, Y.F.; Cui, H.L.; Zhou, R.; Li, L.; Duan, G.L.; Zhu, Y.G. Distinctive Structure and Assembly of Phyllosphere Microbial Communities between Wild and Cultivated Rice. Microbiol. Spectr. 2023, 11, e04371-22. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Singh, B.K.; He, J.Z.; Han, Y.L.; Li, P.P.; Wan, L.H.; Meng, G.Z.; Liu, S.Y.; Wang, J.T.; Wu, C.F.; et al. Plant developmental stage drives the differentiation in ecological role of the maize microbiome. Microbiome 2021, 9, 171. [Google Scholar] [CrossRef]
- Gao, J.; Feng, P.; Zhang, J.; Dong, C.; Wang, Z.; Chen, M.; Yu, Z.; Zhao, B.; Hou, X.; Wang, H.; et al. Enhancing maize’s nitrogen-fixing potential through ZmSBT3, a gene suppressing mucilage secretion. J. Integr. Plant Biol. 2023, 65, 2645–2659. [Google Scholar] [CrossRef]
- Bloch, S.E.; Clark, R.; Gottlieb, S.S.; Wood, L.K.; Shah, N.; Mak, S.M.; Lorigan, J.G.; Johnson, J.; Davis-Richardson, A.G.; Williams, L.; et al. Biological nitrogen fixation in maize: Optimizing nitrogenase expression in a root-associated diazotroph. J. Exp. Bot. 2020, 71, 4591–4603. [Google Scholar] [CrossRef]
- He, X.; Xiao, W.; Zeng, J.; Tang, J.; Wang, L. Detoxification and removal of arsenite by Pseudomonas sp. SMS11: Oxidation, biosorption and bioaccumulation. J. Environ. Manag. 2023, 336, 117641. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, W.; Chen, L.; Zhang, P.; Liu, Z.; Yang, X.; Shao, J.; Ding, Y.; Mi, Y. Effects of pepper-maize intercropping on the physicochemical properties, microbial communities, and metabolites of rhizosphere and bulk soils. Environ. Microbiome 2024, 19, 108. [Google Scholar] [CrossRef]
- Hameed, A.; Nguyen, D.H.; Lin, S.Y.; Stothard, P.; Neelakandan, P.; Young, L.S.; Young, C.C. Hormesis of glyphosate on ferulic acid metabolism and antifungal volatile production in rice root biocontrol endophyte Burkholderia cepacia LS-044. Chemosphere 2023, 345, 140511. [Google Scholar] [CrossRef]
- Hameed, A.; Shahina, M.; Lai, W.A.; Stothard, P.; Young, L.S.; Lin, S.Y.; Young, C.C. Draft genome sequence reveals co-occurrence of multiple antimicrobial resistance and plant probiotic traits in rice root endophytic strain Burkholderia sp. LS-044 affiliated to Burkholderia cepacia complex. J. Glob. Antimicrob. Resist. 2020, 20, 28–30. [Google Scholar] [CrossRef]
- Ikeda, S.; Okubo, T.; Takeda, N.; Banba, M.; Sasaki, K.; Imaizumi-Anraku, H.; Fujihara, S.; Ohwaki, Y.; Ohshima, K.; Fukuta, Y.; et al. The genotype of the calcium/calmodulin-dependent protein kinase gene (CCaMK) determines bacterial community diversity in rice roots under paddy and upland field conditions. Appl. Environ. Microbiol. 2011, 77, 4399–4405. [Google Scholar] [CrossRef]
- Tsotetsi, T.; Nephali, L.; Malebe, M.; Tugizimana, F. Bacillus for Plant Growth Promotion and Stress Resilience: What Have We Learned? Plants 2022, 11, 2482. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, A.; Qin, M.; Elahie, M.; Naeem, M.; Bashir, T.; Yasmin, H.; Younas, M.; Areeb, A.; Irfan, M.; Billah, M.; et al. Bacillus pumilus induced tolerance of Maize (Zea mays L.) against Cadmium (Cd) stress. Sci. Rep. 2021, 11, 17196. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zeng, W.; Ao, C.; Huang, J. Integrative analysis of the transcriptome and metabolome reveals Bacillus atrophaeus WZYH01-mediated salt stress mechanism in maize (Zea mays L.). J. Biotechnol. 2024, 383, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Z.; Pu, Y.; Zhang, B.; Wang, B.; Xing, L.; Li, Y.; Zhang, Y.; Gu, R.; Jia, F.; et al. Antagonistic Strain Bacillus velezensis JZ Mediates the Biocontrol of Bacillus altitudinis m-1, a Cause of Leaf Spot Disease in Strawberry. Int. J. Mol. Sci. 2024, 25, 8872. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, Y.; Jia, T.; Yu, Y.; Chen, S.; Qiu, Y.; Zhang, R.; Chen, H. Controlling mildew of tobacco leaf by Bacillus amyloliquefaciens ZH-2 and its effect on storage quality of tobacco leaf. Sci. Rep. 2025, 15, 5304. [Google Scholar] [CrossRef]
- Chen, S.; Kang, Z.; Peralta-Videa, J.R.; Zhao, L. Environmental implication of MoS(2) nanosheets: Effects on maize plant growth and soil microorganisms. Sci. Total Environ. 2023, 860, 160362. [Google Scholar] [CrossRef]
- Zhao, S.; Gao, N.; Zhang, Q.; Xiao, W.; Chen, D.; Huang, M.; Ye, X. Cultivar-specific rhizosphere microbial community responses to cadmium-NaHCO(3) stress in relation to cadmium accumulation in rice. J. Hazard. Mater. 2025, 488, 137531. [Google Scholar] [CrossRef]
- De la Cruz-Barrón, M.; Cruz-Mendoza, A.; Navarro-Noya, Y.E.; Ruiz-Valdiviezo, V.M.; Ortíz-Gutiérrez, D.; Ramírez-Villanueva, D.A.; Luna-Guido, M.; Thierfelder, C.; Wall, P.C.; Verhulst, N.; et al. The Bacterial Community Structure and Dynamics of Carbon and Nitrogen when Maize (Zea mays L.) and Its Neutral Detergent Fibre Were Added to Soil from Zimbabwe with Contrasting Management Practices. Microb. Ecol. 2017, 73, 135–152. [Google Scholar] [CrossRef]
- Chu, T.N.; Bui, L.V.; Hoang, M.T.T. Pseudomonas PS01 Isolated from Maize Rhizosphere Alters Root System Architecture and Promotes Plant Growth. Microorganisms 2020, 8, 471. [Google Scholar] [CrossRef]
- Gu, Q.; Qiao, J.; Wang, R.; Lu, J.; Wang, Z.; Li, P.; Zhang, L.; Ali, Q.; Khan, A.R.; Gao, X.; et al. The Role of Pyoluteorin from Pseudomonas protegens Pf-5 in Suppressing the Growth and Pathogenicity of Pantoea ananatis on Maize. Int. J. Mol. Sci. 2022, 23, 6431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, Y.N.; Wang, X.; Liao, K.; He, S.; Zhao, X.; Guo, H.; Zhao, D.; Wei, H.L. Dynamics of rice microbiomes reveal core vertically transmitted seed endophytes. Microbiome 2022, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil. 2018, 422, 7–34. [Google Scholar] [CrossRef]
- Chowhan, P.; Swarnakar, S.; Chakraborty, A.P. New report of endophytic bacterium Achromobacter xylosoxidans from root tissue of Musa spp. Mol. Biol. Rep. 2023, 50, 9179–9190. [Google Scholar] [CrossRef]
- Ely, C.S.; Smets, B.F. Guild Composition of Root-Associated Bacteria Changes with Increased Soil Contamination. Microb. Ecol. 2019, 78, 416–427. [Google Scholar] [CrossRef]
- Jiménez-Vázquez, K.R.; García-Cárdenas, E.; Barrera-Ortiz, S.; Ortiz-Castro, R.; Ruiz-Herrera, L.F.; Ramos-Acosta, B.P.; Coria-Arellano, J.L.; Sáenz-Mata, J.; López-Bucio, J. The plant beneficial rhizobacterium Achromobacter sp. 5B1 influences root development through auxin signaling and redistribution. Plant J. 2020, 103, 1639–1654. [Google Scholar] [CrossRef]
- Kryuchkova, Y.V.; Neshko, A.A.; Gogoleva, N.E.; Balkin, A.S.; Safronova, V.I.; Kargapolova, K.Y.; Shagimardanova, E.I.; Gogolev, Y.V.; Burygin, G.L. Genomics and taxonomy of the glyphosate-degrading, copper-tolerant rhizospheric bacterium Achromobacter insolitus LCu2. Antonie Van Leeuwenhoek 2024, 117, 105. [Google Scholar] [CrossRef]
- Kour, D.; Kour, H.; Khan, S.S.; Khan, R.T.; Bhardwaj, M.; Kailoo, S.; Kumari, C.; Rasool, S.; Yadav, A.N.; Sharma, Y.P. Biodiversity and Functional Attributes of Rhizospheric Microbiomes: Potential Tools for Sustainable Agriculture. Curr. Microbiol. 2023, 80, 192. [Google Scholar] [CrossRef]
- Blake, C.; Christensen, M.N.; Kovács Á, T. Molecular Aspects of Plant Growth Promotion and Protection by Bacillus subtilis. Mol. Plant Microbe Interact. 2021, 34, 15–25. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, X.; Lang, D.; Zhang, X.; Zhou, L.; Wang, L.; Zhang, X. Encapsulation of Bacillus pumilus G5 from polyvinyl alcohol-sodium alginate (PVA-SA) and its implications in improving plant growth and soil fertility under drought and salt soil conditions. Int. J. Biol. Macromol. 2022, 209, 231–243. [Google Scholar] [CrossRef]
- Zaid, D.S.; Li, W.; Yang, S.; Li, Y. Identification of bioactive compounds of Bacillus velezensis HNA3 that contribute to its dual effects as plant growth promoter and biocontrol against post-harvested fungi. Microbiol. Spectr. 2023, 11, e0051923. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Chen, Q.; White, J.F. Evaluation of colonization and mutualistic endophytic symbiosis of Escherichia coli with tomato and Bermuda grass seedlings. PeerJ 2022, 10, e13879. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.A.; Tariq, L.; Nissar, S.; Islam, S.T.; Islam, S.U.; Mangral, Z.; Ilyas, N.; Sayyed, R.Z.; Muthusamy, G.; Kim, W.; et al. The role of plant-associated rhizobacteria in plant growth, biocontrol and abiotic stress management. J. Appl. Microbiol. 2022, 133, 2717–2741. [Google Scholar] [CrossRef]
- Cherif-Silini, H.; Thissera, B.; Bouket, A.C.; Saadaoui, N.; Silini, A.; Eshelli, M.; Alenezi, F.N.; Vallat, A.; Luptakova, L.; Yahiaoui, B.; et al. Durum Wheat Stress Tolerance Induced by Endophyte Pantoea agglomerans with Genes Contributing to Plant Functions and Secondary Metabolite Arsenal. Int. J. Mol. Sci. 2019, 20, 3989. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.F.; Barbosa, R.R.; de Souza, A.N.; da Motta, O.V.; Teixeira, G.N.; Carvalho, V.S.; de Souza, A.L.; de Souza Filho, G.A. Isolation of Pantoea ananatis from sugarcane and characterization of its potential for plant growth promotion. Genet. Mol. Res. 2015, 14, 15301–15311. [Google Scholar] [CrossRef]
- Niu, H.; Sun, Y.; Zhang, Z.; Zhao, D.; Wang, N.; Wang, L.; Guo, H. The endophytic bacterial entomopathogen Serratia marcescens promotes plant growth and improves resistance against Nilaparvata lugens in rice. Microbiol. Res. 2022, 256, 126956. [Google Scholar] [CrossRef]
- Mondal, M.; Kumar, V.; Bhatnagar, A.; Vithanage, M.; Selvasembian, R.; Ambade, B.; Meers, E.; Chaudhuri, P.; Biswas, J.K. Bioremediation of metal(loid) cocktail, struvite biosynthesis and plant growth promotion by a versatile bacterial strain Serratia sp. KUJM3: Exploiting environmental co-benefits. Environ. Res. 2022, 214, 113937. [Google Scholar] [CrossRef]
- Chopra, A.; Mongad, D.; Satpute, S.; Mazumder, P.B.; Rahi, P. Quorum sensing activities and genomic insights of plant growth-promoting rhizobacteria isolated from Assam tea. World. J. Microbiol. Biotechnol. 2023, 39, 160. [Google Scholar] [CrossRef]
- Martin, P.A.W.; Gundersen-Rindal, D.; Blackburn, M.; Buyer, J. Chromobacterium subtsugae sp. nov., a betaproteobacterium toxic to Colorado potato beetle and other insect pests. Int. J. Syst. Evol. Microbiol. 2007, 57, 993–999. [Google Scholar] [CrossRef]
- Wang, P.; Wei, H.; Ke, T.; Fu, Y.; Zeng, Y.; Chen, C.; Chen, L. Characterization and genome analysis of Acinetobacter oleivorans S4 as an efficient hydrocarbon-degrading and plant-growth-promoting rhizobacterium. Chemosphere 2023, 331, 138732. [Google Scholar] [CrossRef]
- Shah, Z.M.; Naz, R.; Naz, S.; Zahoor, S.; Nosheen, A.; Shahid, M.; Anwar, Z.; Keyani, R. Incorporation of zinc sulfide nanoparticles, Acinetobacter pittii and Bacillus velezensis to improve tomato plant growth, biochemical attributes and resistance against Rhizoctoniasolani. Plant Physiol. Biochem. 2023, 202, 107909. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Roychoudhury, A. Bio-priming with a Novel Plant Growth-Promoting Acinetobacter indicus Strain Alleviates Arsenic-Fluoride Co-toxicity in Rice by Modulating the Physiome and Micronutrient Homeostasis. Appl. Biochem. Biotechnol. 2023, 195, 6441–6464. [Google Scholar] [CrossRef] [PubMed]
- Samaddar, S.; Chatterjee, P.; Roy Choudhury, A.; Ahmed, S.; Sa, T. Interactions between Pseudomonas spp. and their role in improving the red pepper plant growth under salinity stress. Microbiol Res. 2019, 219, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Torres, P.; Cárdenas, S.; Arismendi Macuer, M.; Huanacuni, N.; Huanca-Mamani, W.; Cifuentes, D.; Sepúlveda Chavera, G.F. The Endophytic Pseudomonas sp. S57 for Plant-Growth Promotion and the Biocontrol of Phytopathogenic Fungi and Nematodes. Plants 2021, 10, 1531. [Google Scholar] [CrossRef]
- Majeed, A.; Kaleem Abbasi, M.; Hameed, S.; Yasmin, S.; Hanif, M.K.; Naqqash, T.; Imran, A. Pseudomonas sp. AF-54 containing multiple plant beneficial traits acts as growth enhancer of Helianthus annuus L. under reduced fertilizer input. Microbiol. Res. 2018, 216, 56–69. [Google Scholar] [CrossRef]
- de Matos, G.F.; Rouws, L.F.M.; Simões-Araújo, J.L.; Baldani, J.I. Evolution and function of nitrogen fixation gene clusters in sugarcane associated Bradyrhizobium strains. Environ. Microbiol. 2021, 23, 6148–6162. [Google Scholar] [CrossRef]
- Hashimoto, S.; Goto, K.; Pyromyou, P.; Songwattana, P.; Greetatorn, T.; Tittabutr, P.; Boonkerd, N.; Teaumroong, N.; Uchiumi, T. Type III Secretion System of Bradyrhizobium sp. SUTN9-2 Obstructs Symbiosis with Lotus spp. Microbes Environ. 2020, 35. [Google Scholar] [CrossRef]
- Balázs, H.E.; Schmid, C.A.O.; Cruzeiro, C.; Podar, D.; Szatmari, P.M.; Buegger, F.; Hufnagel, G.; Radl, V.; Schröder, P. Post-reclamation microbial diversity and functions in hexachlorocyclohexane (HCH) contaminated soil in relation to spontaneous HCH tolerant vegetation. Sci. Total Environ. 2021, 767, 144653. [Google Scholar] [CrossRef]
- Wang, F.; Wei, Y.; Yan, T.; Wang, C.; Chao, Y.; Jia, M.; An, L.; Sheng, H. Sphingomonas sp. Hbc-6 alters physiological metabolism and recruits beneficial rhizosphere bacteria to improve plant growth and drought tolerance. Front. Plant Sci. 2022, 13, 1002772. [Google Scholar] [CrossRef]
- Timilsina, S.; Potnis, N.; Newberry, E.A.; Liyanapathiranage, P.; Iruegas-Bocardo, F.; White, F.F.; Goss, E.M.; Jones, J.B. Xanthomonas diversity, virulence and plant-pathogen interactions. Nat. Rev. Microbiol. 2020, 18, 415–427. [Google Scholar] [CrossRef]
- Jacques, M.A.; Arlat, M.; Boulanger, A.; Boureau, T.; Carrère, S.; Cesbron, S.; Chen, N.W.; Cociancich, S.; Darrasse, A.; Denancé, N.; et al. Using Ecology, Physiology, and Genomics to Understand Host Specificity in Xanthomonas. Annu. Rev. Phytopathol. 2016, 54, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 2023, 51, D18–D28. [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Peng, Q.; Chen, S.; Liu, Z.; Zhang, W.; Zhang, C.; Du, X.; Sun, S.; Peng, W.; Lei, Z.; et al. Microbiome Migration from Soil to Leaves in Maize and Rice. Microorganisms 2025, 13, 947. https://doi.org/10.3390/microorganisms13040947
Ma J, Peng Q, Chen S, Liu Z, Zhang W, Zhang C, Du X, Sun S, Peng W, Lei Z, et al. Microbiome Migration from Soil to Leaves in Maize and Rice. Microorganisms. 2025; 13(4):947. https://doi.org/10.3390/microorganisms13040947
Chicago/Turabian StyleMa, Jiejia, Qianze Peng, Silu Chen, Zhuoxin Liu, Weixing Zhang, Chi Zhang, Xiaohua Du, Shue Sun, Weiye Peng, Ziling Lei, and et al. 2025. "Microbiome Migration from Soil to Leaves in Maize and Rice" Microorganisms 13, no. 4: 947. https://doi.org/10.3390/microorganisms13040947
APA StyleMa, J., Peng, Q., Chen, S., Liu, Z., Zhang, W., Zhang, C., Du, X., Sun, S., Peng, W., Lei, Z., Zhang, L., Su, P., Zhang, D., & Liu, Y. (2025). Microbiome Migration from Soil to Leaves in Maize and Rice. Microorganisms, 13(4), 947. https://doi.org/10.3390/microorganisms13040947