
Transcriptomic Insights into the Degradation Mechanisms of Fomitopsis pinicola and Its Host Preference for Coniferous over Broadleaf Deadwood


Abstract
:1. Introduction
2. Materials and Methods
2.1. Degradation Experiment Setup
2.2. Weight, Cellulose, and Lignin Loss of Wooden Strips After Degradation
2.3. RNA Extraction, Library Construction, and RNA-Seq
2.4. Data Processing and Visualization Analysis
3. Results
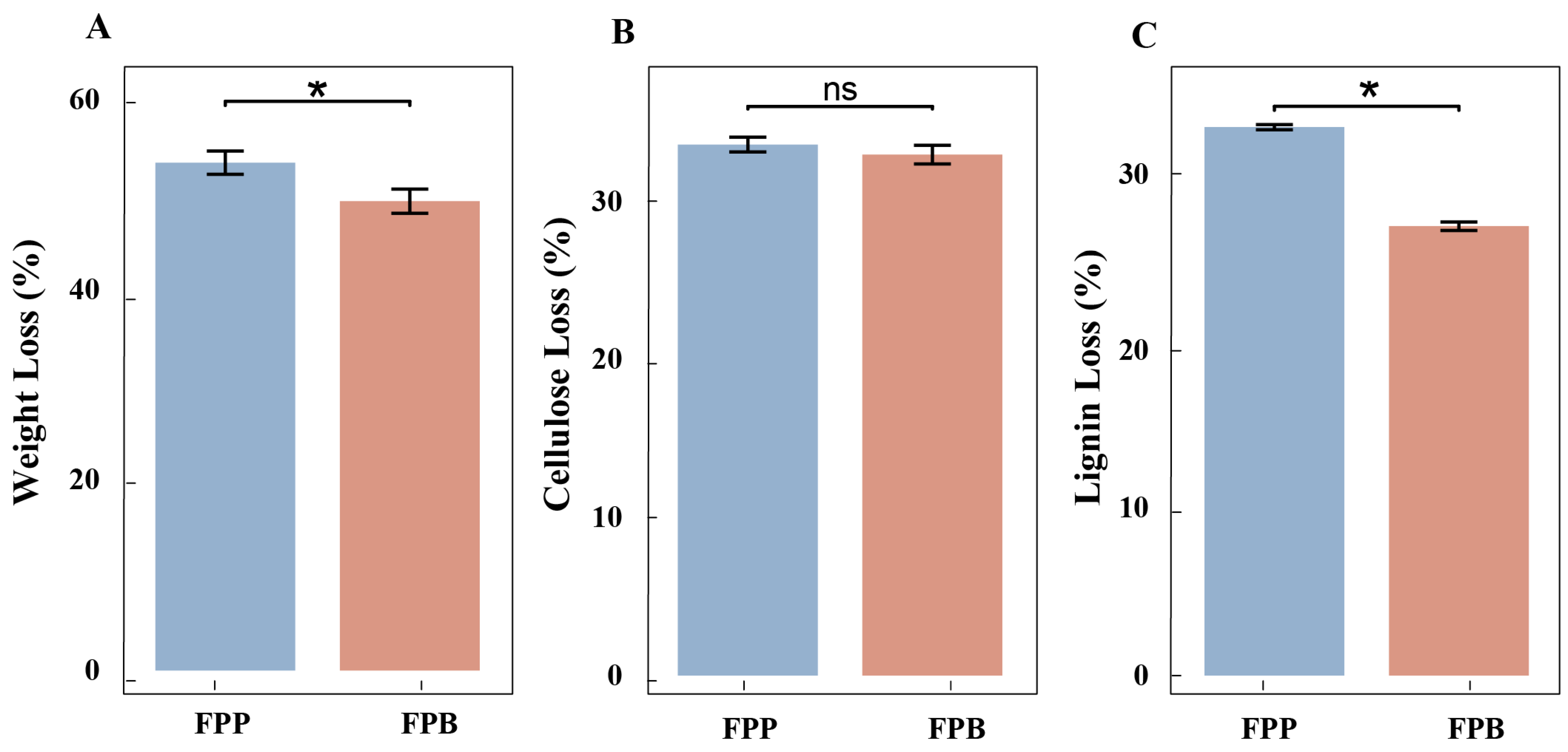
3.1. Wooden Strip Degradation
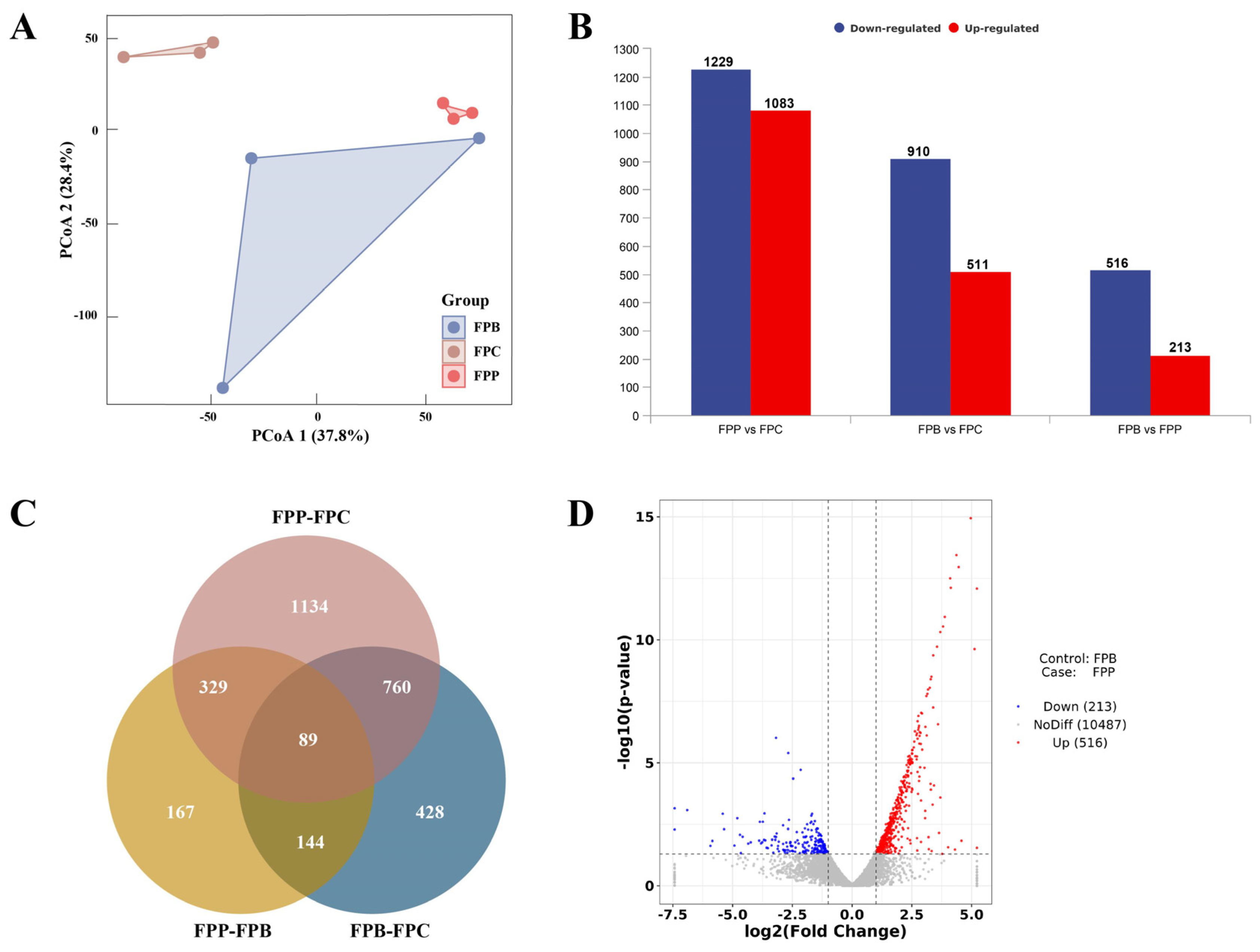
3.2. Data Quality Control Analysis of Transcriptome Sequencing
3.3. Analysis of Differential Expression of mRNA
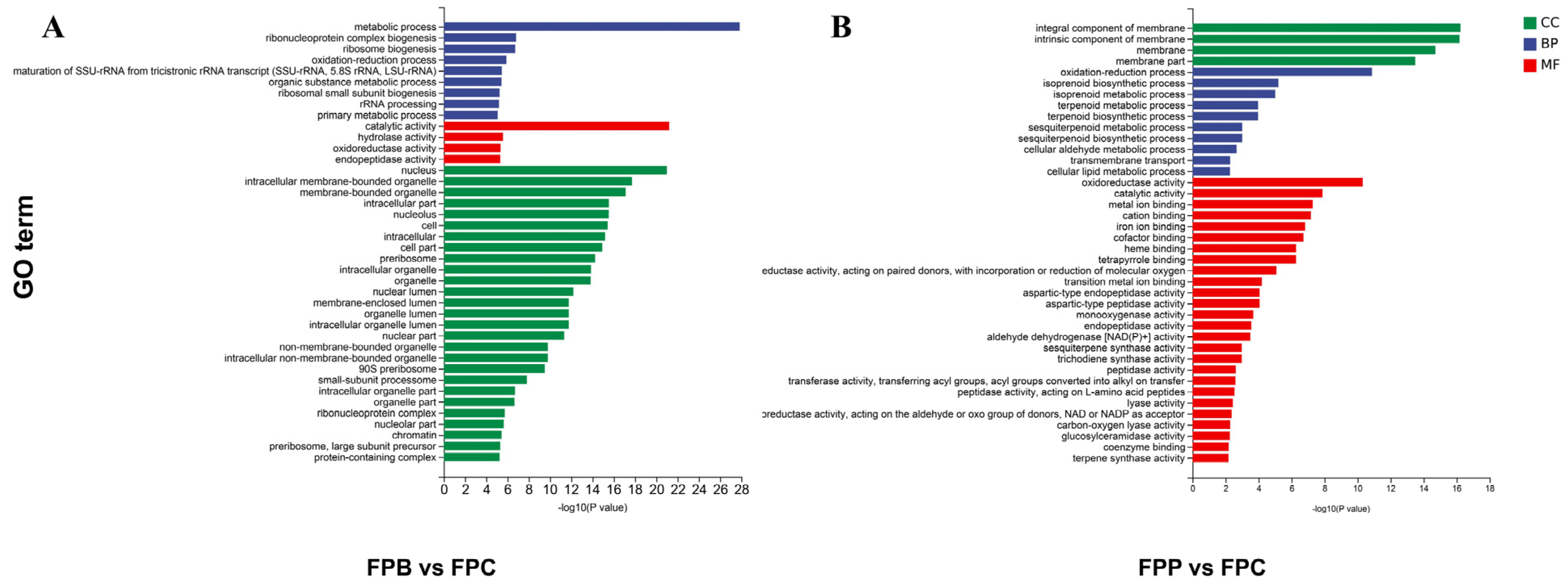
3.4. Functional Annotation of Differentially Expressed Genes
3.5. Analysis of Different Genes Using KEGG
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harmon, M.; Franklin, J.; Swanson, F.; Sollins, P.; Gregory, S.; Lattin, J.; Anderson, N. Ecology of coarse wooden debris in temperate ecosystems. Adv. Ecol. Res. 1986, 15, 133–302. [Google Scholar]
- Stokland, J.; Siitonen, J.; Jonsson, B. Biodiversity in Dead Wood; Cambridge University Press: New York, NY, USA, 2012. [Google Scholar]
- Oberle, B.; Ogle, K.; Zanne, A.E.; Woodall, C.W. When a tree falls: Controls on wood decay predict standing dead tree fall and new risks in Changing forests. PLoS ONE 2018, 13, e0196712. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1871–1960. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef]
- Purahong, W.; Wubet, T.; Krüger, D.; Buscot, F. Molecular evidence strongly supports deadwood-inhabiting fungi exhibiting unexpected tree species preferences in temperate forests. ISME J. 2018, 12, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Rajala, T.; Peltoniemi, M.; Pennanen, T.; Mäkipää, R. Fungal community dynamics in relation to substrate quality of decaying Norway spruce (Picea abies [L.] Karst.) logs in boreal forests. FEMS Microbiol. Ecol. 2012, 81, 494–505. [Google Scholar] [CrossRef]
- Hori, C.; Gaskell, J.; Igarashi, K.; Samejima, M.; Hibbett, D.; Henrissat, B.; Cullen, D. Genomewide analysis of polysaccharides degrading enzymes in 11 white- and brown-rot Polyporales provides insight into mechanisms of wood decay. Mycologia 2013, 105, 1412–1427. [Google Scholar] [CrossRef]
- Goodell, B.; Jellison, J.; Liu, J.; Daniel, G.; Paszczynski, A.; Fekete, F.; Krishnamurthy, S.; Jun, L.; Xu, G. Low molecular weight chelators and phenolic compounds isolated from wood decay fungi and their role in the fungal biodegradation of wood. J. Biotechnol. 1997, 53, 133–162. [Google Scholar] [CrossRef]
- Paszczynski, A.; Crawford, R.; Funk, D.; Goodell, B. De Novo synthesis of 4,5-Dimethoxycatechol and 2,5-Dimethoxyhydroquinone by the brown rot fungus Gloeophyllum trabeum. Appl. Environ. Microb. 1999, 65, 674–679. [Google Scholar] [CrossRef]
- Jensen, K.A.; Houtman, C.J.; Ryan, Z.C.; Hammel, K.E. Pathways for extracellular Fenton chemistry in the brown rot Basidiomycete Gloeophyllum trabeum. Appl. Environ. Microb. 2001, 67, 2705–2711. [Google Scholar] [CrossRef]
- Lynch, M.; Gabriel, W. Environmental tolerance. Am. Nat. 1987, 129, 283–303. [Google Scholar] [CrossRef]
- Sexton, J.P.; Montiel, J.; Shay, J.E.; Stephens, M.R.; Slatyer, R.A. Evolution of ecological niche breadth. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 183–206. [Google Scholar] [CrossRef]
- Krah, F.-S.; Bässler, C.; Heibl, C.; Soghigian, J.; Schaefer, H.; Hibbett, D.S. Evolutionary dynamics of host specialization in wood-decay fungi. BMC Ecol. Evol. 2018, 18, 119. [Google Scholar] [CrossRef]
- Bütler, R.; Patty, L.; Le Bayon, R.-C.; Guenat, C.; Schlaepfer, R. Log decay of Picea Abies in the Swiss Jura mountains of central Europe. For. Ecol. Manag. 2007, 242, 791–799. [Google Scholar] [CrossRef]
- Han, M.-L.; Chen, Y.-Y.; Shen, L.-L.; Song, J.; Vlasák, J.; Dai, Y.-C.; Cui, B.-K. Taxonomy and phylogeny of the brown-rot fungi: Fomitopsis and its related genera. Fungal Divers. 2016, 80, 343–373. [Google Scholar] [CrossRef]
- Liu, S.; Shen, L.L.; Wang, Y.; Xu, T.M.; Gates, G.; Cui, B.K. Species diversity and molecular phylogeny of Cyanosporus (Polyporales, Basidiomycota). Front. Microbiol. 2021, 12, 631166. [Google Scholar] [CrossRef]
- Hao, Z.Q.; Li, B.H.; Zhang, J.; Wang, X.G.; Ye, J.; Yao, X. Broad-leaved korean pine (Pinus koraiensis) mixed forest plot in changbai mountain (CBS) of China: Community composition and structure. Chin. J. Plant Ecol. 2008, 32, 238–250. [Google Scholar]
- Hao, Z.Q.; Zhang, J.; Li, B.H.; Ye, J.; Wang, X.G.; Yao, X. Natural secondary poplar-birch forest in changbai mountain: Species composition and community structure. Chin. J. Plant Ecol. 2008, 32, 251–261. [Google Scholar]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef]
- Cloonan, N.; Forrest, A.R.R.; Kolle, G.; Gardiner, B.B.A.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ahmad, M.; Abdul Aziz, M.; Sabeem, M.; Kutty, M.S.; Sivasankaran, S.K.; Brini, F.; Xiao, T.T.; Blilou, I.; Masmoudi, K. Date palm transcriptome analysis provides new insights on changes in response to high salt stress of colonized roots with the endophytic fungus piriformospora indica. Front. Plant Sci. 2024, 15, 1400215. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; He, K.; Li, M.; Zhang, Y.; Jiang, J.; Qian, L.; Gao, X.; Zhang, C.; Liu, S. Comparative transcriptome analysis of Fusarium graminearum challenged with distinct fungicides and functional analysis of FgICL gene. Genomics 2024, 116, 110869. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.L.; Xue, J.B.; Shi, J.T.; Li, T.; Yuan, H.S. Comparative transcriptome analysis explores the mechanism of angiosperm and gymnosperm deadwood degradation by Fomes fomentarius. J. Fungi 2024, 10, 196. [Google Scholar] [CrossRef]
- Yang, H.; Wang, C.; Zhong, S.; Yang, H.; Chen, C.; Tan, F.; Ren, T.; Shen, J.; Luo, P. Identification of pathogenic-like fungal species on Akebia trifoliata fruit by integrating transcriptome analysis and specific sequences of putative pathogens. Physiol. Mol. Plant Pathol. 2024, 132, 102297. [Google Scholar] [CrossRef]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Hatakka, A.; Hammel, K.E. Fungal biodegradation of lignocelluloses. In Industrial Applications; Hofrichter, M., Ed.; The Mycota; Springer: Berlin, Germany, 2011; Volume 10, pp. 319–340. [Google Scholar]
- Lundell, T.K.; Mäkelä, M.R.; Hildén, K. Lignin-modifying enzymes in filamentous basidiomycetes—Ecological, functional and phylogenetic review. J. Basic Microbiol. 2010, 50, 5–20. [Google Scholar] [CrossRef]
- Sista Kameshwar, A.K.; Qin, W. Systematic metadata analysis of brown rot fungi gene expression data reveals the genes involved in Fenton’s reaction and wood decay process. Mycology 2020, 11, 22–37. [Google Scholar] [CrossRef]
- Schwarze, F.W.M.R. Wood decay under the microscope. Fungal Biol. Rev. 2007, 21, 133–170. [Google Scholar] [CrossRef]
- Liu, S.; Song, C.G.; Xu, T.M.; Ji, X.; Wu, D.M.; Cui, B.K. Species diversity, molecular phylogeny, and ecological habits of Fomitopsis (Polyporales, Basidiomycota). Front. Microbiol. 2022, 13, 859411. [Google Scholar] [CrossRef]
- Haight, J.E.; Nakasone, K.K.; Laursen, G.A.; Redhead, S.A.; Taylor, D.L.; Glaeser, J.A. Fomitopsis mounceae and F. schrenkii—Two new species from North America in the F. pinicola complex. Mycologia 2019, 111, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, X.; Li, S.; Wei, B. Lignin and cellulose contents in Chinese red pine (Pinus tabuliformis Carr.) plantations varied in stand structure, soil property, and regional climate. Forests 2024, 15, 240. [Google Scholar] [CrossRef]
- Pan, Y.; Qin, R.; Hou, M.; Xue, J.; Zhou, M.; Xu, L.; Zhang, Y. The interactions of polyphenols with Fe and their application in Fenton/Fenton-like reactions. Sep. Purif. Technol. 2022, 300, 121831. [Google Scholar] [CrossRef]
- Rehbein, M.; Koch, G. Topochemical investigation of early stages of lignin modification within individual cell wall layers of Scots Pine (Pinus sylvestris L.) sapwood infected by the brown-rot fungus Antrodia Vaillantii (DC.: Fr.) Ryv. Int. Biodeterior. Biodegrad. 2011, 65, 913–920. [Google Scholar] [CrossRef]
- Arnstadt, T.; Hoppe, B.; Kahl, T.; Kellner, H.; Krüger, D.; Bässler, C.; Bauhus, J.; Hofrichter, M. Patterns of laccase and peroxidases in coarse woody debris of Fagus sylvatica, Picea abies and Pinus sylvestris and their relation to different wood parameters. Eur. J. For. Res. 2016, 135, 109–124. [Google Scholar] [CrossRef]
- De Figueiredo, F.L.; De Oliveira, A.C.P.; Terrasan, C.R.F.; Gonçalves, T.A.; Gerhardt, J.A.; Tomazetto, G.; Persinoti, G.F.; Rubio, M.V.; Peña, J.A.T.; Araújo, M.F.; et al. Multi-omics analysis provides insights into lignocellulosic biomass degradation by Laetiporus sulphureus ATCC 52600. Biotechnol. Biofuels Bioprod. 2021, 14, 96. [Google Scholar] [CrossRef]
- Csarman, F.; Obermann, T.; Zanjko, M.C.; Man, P.; Halada, P.; Seiboth, B.; Ludwig, R. Functional expression and characterization of two laccases from the brown rot Fomitopsis pinicola. Enzym. Microb. Technol. 2021, 148, 109801. [Google Scholar] [CrossRef]
- Park, N.; Park, S. Purification and characterization of a novel laccase from Fomitopsis pinicola mycelia. Int. J. Biol. Macromol. 2014, 70, 583–589. [Google Scholar] [CrossRef]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- Dai, Z.; Shestov, A.A.; Lai, L.; Locasale, J.W. A flux balance of glucose metabolism clarifies the requirements of the Warburg effect. Biophys. J. 2016, 111, 1088–1100. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, Y.; Li, Y.; Wu, X.; Chen, S.; Wang, M.; Li, Y.; Ge, Z.; Zhang, M.; Mao, L. The evolutionary adaptation of wood-decay macrofungi to host gymnosperms differs from that to host angiosperms. Ecol. Evol. 2024, 14, e70019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GO Terms | Upregulated Genes | Downregulated Genes |
|---|---|---|---|
| BP | cellular aldehyde metabolic process cellular lipid metabolic process cellular response to toxic substance detoxification isoprenoid biosynthetic process isoprenoid metabolic process lipid biosynthetic process oxidation-reduction process response to toxic substance transmembrane transport | 162 | 93 |
| CC | cellular anatomical entity integral component of membrane intrinsic component of membrane membrane | 528 | 238 |
| MF | catalytic activity cation binding cofactor binding coenzyme binding heme binding iron ion binding metal ion binding oxidoreductase activity tetrapyrrole binding transition metal ion binding | 684 | 350 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, J.; Wei, Y.; Chen, L.; Yuan, H. Transcriptomic Insights into the Degradation Mechanisms of Fomitopsis pinicola and Its Host Preference for Coniferous over Broadleaf Deadwood. Microorganisms 2025, 13, 1006. https://doi.org/10.3390/microorganisms13051006
Xue J, Wei Y, Chen L, Yuan H. Transcriptomic Insights into the Degradation Mechanisms of Fomitopsis pinicola and Its Host Preference for Coniferous over Broadleaf Deadwood. Microorganisms. 2025; 13(5):1006. https://doi.org/10.3390/microorganisms13051006
Chicago/Turabian StyleXue, Jianbin, Yulian Wei, Liting Chen, and Haisheng Yuan. 2025. "Transcriptomic Insights into the Degradation Mechanisms of Fomitopsis pinicola and Its Host Preference for Coniferous over Broadleaf Deadwood" Microorganisms 13, no. 5: 1006. https://doi.org/10.3390/microorganisms13051006
APA StyleXue, J., Wei, Y., Chen, L., & Yuan, H. (2025). Transcriptomic Insights into the Degradation Mechanisms of Fomitopsis pinicola and Its Host Preference for Coniferous over Broadleaf Deadwood. Microorganisms, 13(5), 1006. https://doi.org/10.3390/microorganisms13051006