
Multi-Metagenome Analysis Unravels Community Collapse After Sampling and Hints the Cultivation Strategy of CPR Bacteria in Groundwater


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. DNA Extraction, Metagenomic Sequencing, and Assembly
2.3. Bioinformatics Analyses
2.4. Construction of Co-Occurrence Networks
3. Results and Discussion
3.1. Metagenome Sequence Yield and Microbial Community
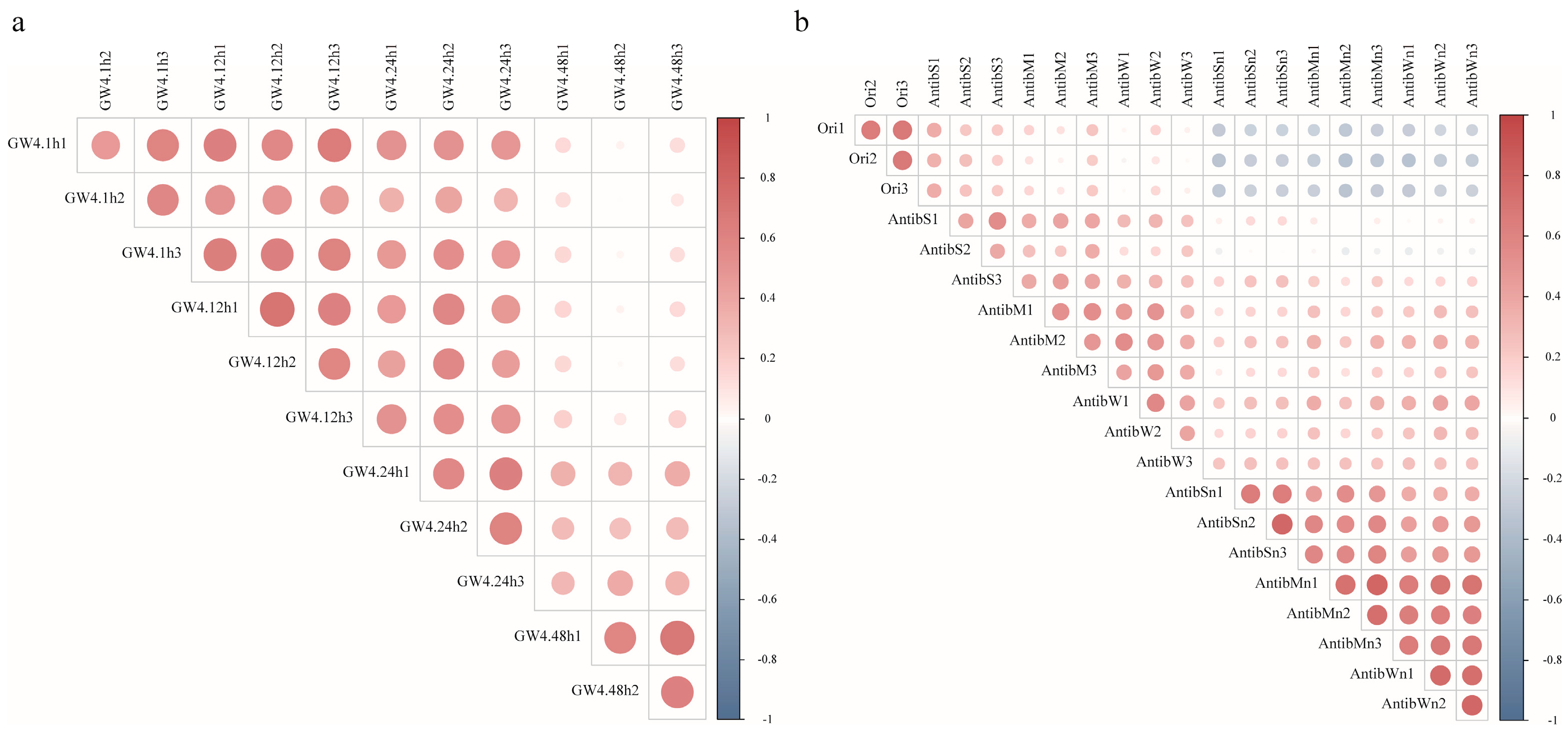
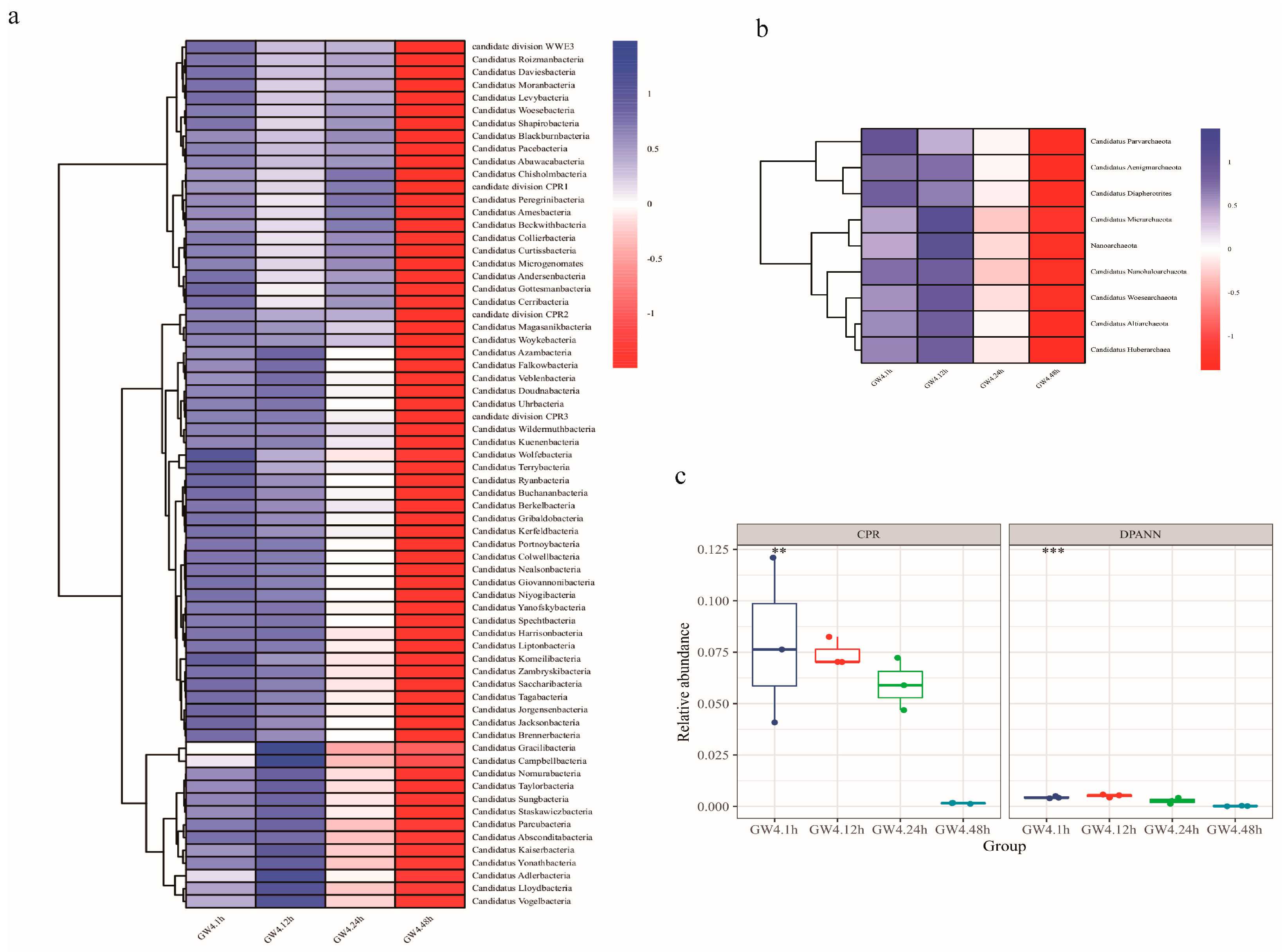
3.2. Temporal Dynamics of CPR Communities Post-Sampling
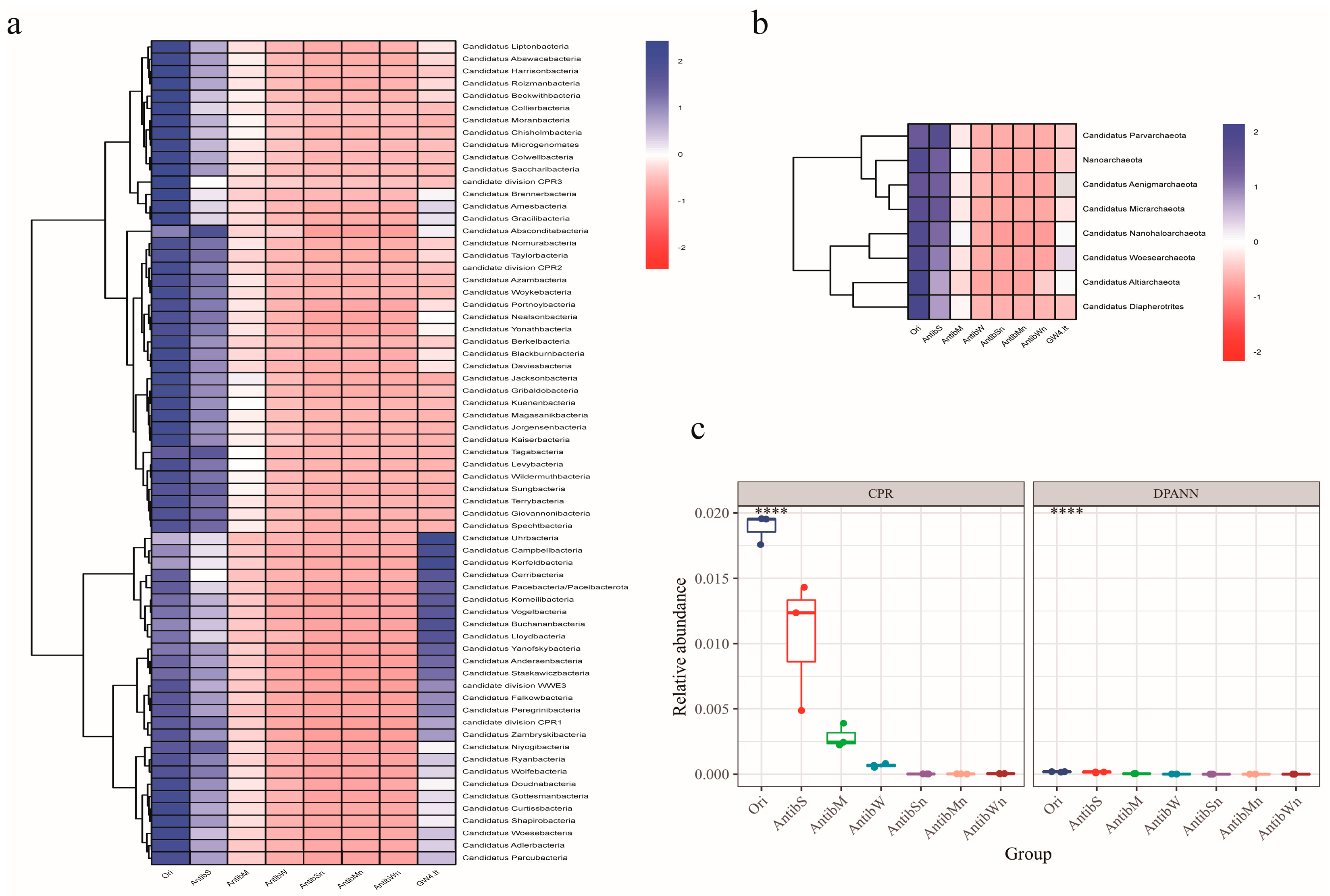
3.3. Ampicillin Suppression and the Nutrient Paradox
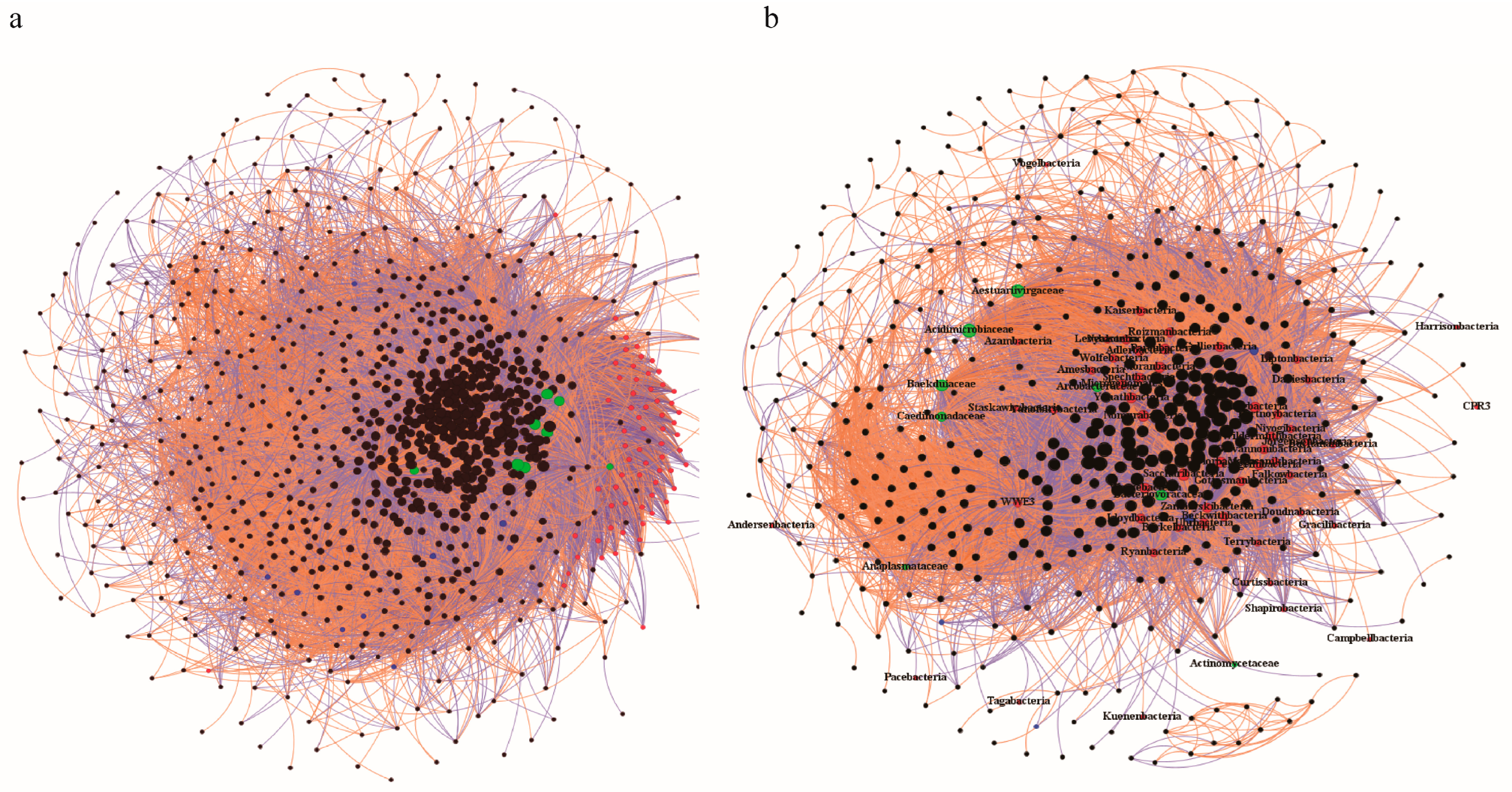
3.4. Construction of Co-Occurrence Networks and Speculation on Potential Host Bacteria of CPR
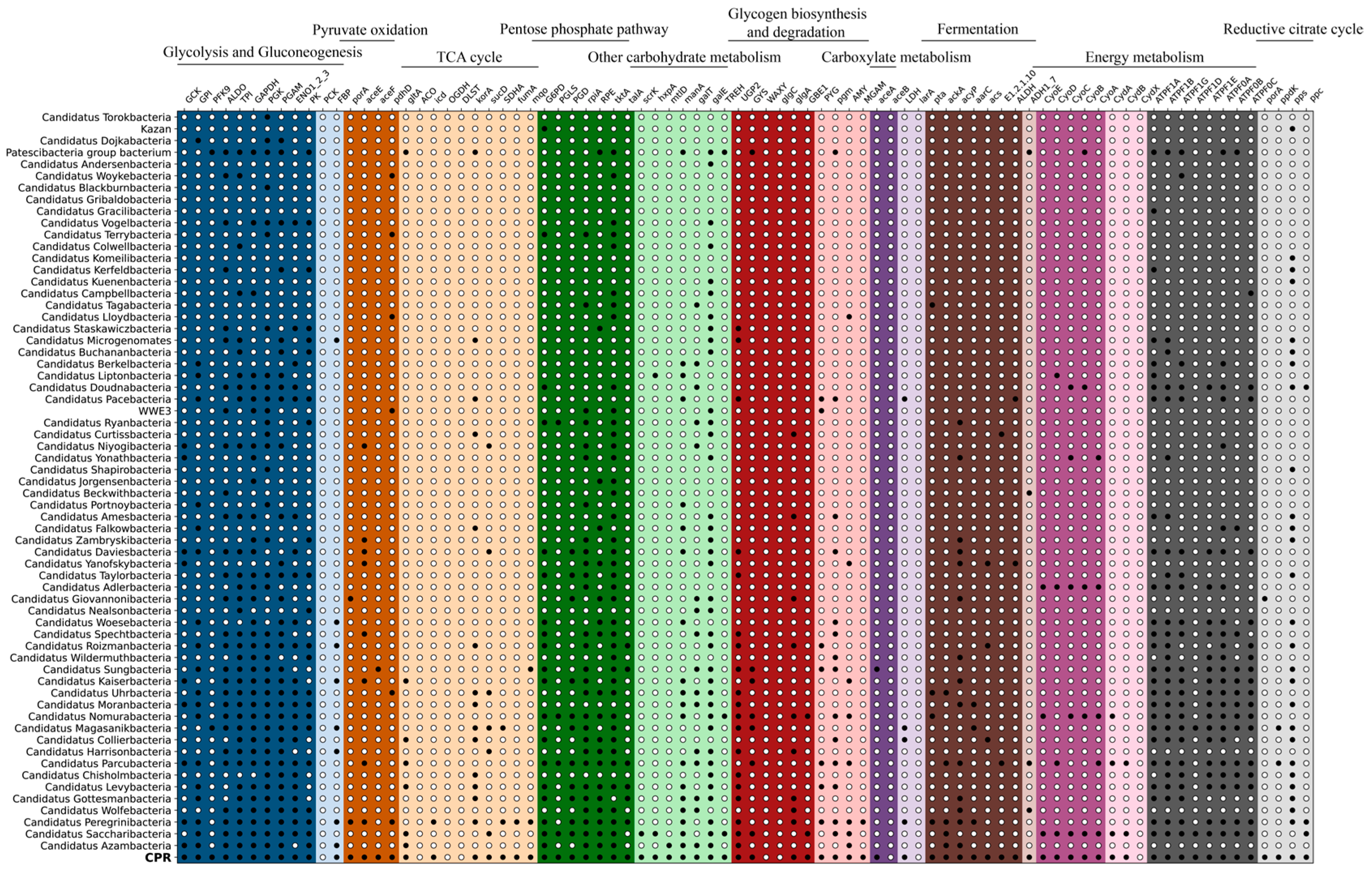
3.5. Metabolic Potential of CPR Bacteria in Groundwater
3.6. Insights into the Cultivation of CPR Bacteria in Groundwater
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef]
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef]
- Harris, J.K.; Kelley, S.T.; Pace, N.R. New perspective on uncultured bacterial phylogenetic division OP11. Appl. Environ. Microbiol. 2004, 70, 845–849. [Google Scholar] [CrossRef]
- Castelle, C.J.; Banfield, J.F. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 2018, 172, 1181–1197. [Google Scholar] [CrossRef]
- Castelle, C.J.; Brown, C.T.; Anantharaman, K.; Probst, A.J.; Huang, R.H.; Banfield, J.F. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 2018, 16, 629–645. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Dutkiewicz, Z.; Singleton, C.M.; Sereika, M.; Villada, J.C.; Mussig, A.J.; Chuvochina, M.; Albertsen, M.; Schulz, F.; Woyke, T.; Nielsen, P.H.; et al. Proposal of Patescibacterium danicum gen. nov., sp. nov. in the ubiquitous bacterial phylum Patescibacteriota phyl. nov. ISME Commun. 2024, 5, ycae147. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Keren, R.; Whittaker, M.L.; Farag, I.F.; Doudna, J.A.; Cate, J.H.D.; Banfield, J.F. Genome-resolved metagenomics reveals site-specific diversity of episymbiotic CPR bacteria and DPANN archaea in groundwater ecosystems. Nat. Microbiol. 2021, 6, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.M.; Jaffe, A.L.; Nuccio, E.E.; Taga, M.E.; Firestone, M.K.; Banfield, J.F. Soil Candidate Phyla Radiation Bacteria Encode Components of Aerobic Metabolism and Co-occur with Nanoarchaea in the Rare Biosphere of Rhizosphere Grassland Communities. mSystems 2021, 6, e0120520. [Google Scholar] [CrossRef]
- Linz, A.M.; Crary, B.C.; Shade, A.; Owens, S.; Gilbert, J.A.; Knight, R.; McMahon, K.D. Bacterial Community Composition and Dynamics Spanning Five Years in Freshwater Bog Lakes. mSphere 2017, 2, e00169-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; Al-Shayeb, B.; Méheust, R.; Li, W.J.; Doudna, J.A.; Banfield, J.F. Candidate Phyla Radiation Roizmanbacteria from hot springs have novel and unexpectedly abundant CRISPR-Cas systems. Front. Microbiol. 2019, 10, 928. [Google Scholar] [CrossRef]
- Zhou, Z.; St John, E.; Anantharaman, K.; Reysenbach, A.L. Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits. Microbiome 2022, 10, 241. [Google Scholar] [CrossRef]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Kristensen, J.M.; Herbold, C.W.; Pjevac, P.; Kitzinger, K.; Hausmann, B.; Dueholm, M.K.D.; Nielsen, P.H.; Wagner, M. Global abundance patterns, diversity, and ecology of Patescibacteria in wastewater treatment plants. Microbiome 2024, 12, 55. [Google Scholar] [CrossRef]
- Bruno, A.; Sandionigi, A.; Bernasconi, M.; Panio, A.; Labra, M.; Casiraghi, M. Changes in the drinking water microbiome: Effects of water treatments along the flow of two drinking water treatment plants in an urbanized area, Milan (Italy). Front. Microbiol. 2018, 9, 2557. [Google Scholar] [CrossRef]
- Masuda, S.; Gan, P.; Kiguchi, Y.; Anda, M.; Sasaki, K.; Shibata, A.; Iwasaki, W.; Suda, W.; Shirasu, K. Uncovering microbiomes of the rice phyllosphere using long-read metagenomic sequencing. Commun. Biol. 2024, 7, 357. [Google Scholar] [CrossRef]
- Naud, S.; Ibrahim, A.; Valles, C.; Maatouk, M.; Bittar, F.; Tidjani Alou, M.; Raoult, D. Candidate Phyla Radiation, an underappreciated division of the human microbiome, and its impact on health and disease. Clin. Microbiol. Rev. 2022, 35, e0014021. [Google Scholar] [CrossRef]
- Srinivas, P.; Peterson, S.B.; Gallagher, L.A.; Wang, Y.; Mougous, J.D. Beyond genomics in Patescibacteria: A trove of unexplored biology packed into ultrasmall bacteria. Proc. Natl. Acad. Sci. USA 2024, 121, e2419369121. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Sandionigi, A.; Rizzi, E.; Bernasconi, M.; Vicario, S.; Galimberti, A.; Cocuzza, C.; Labra, M.; Casiraghi, M. Exploring the under-investigated “microbial dark matter” of drinking water treatment plants. Sci. Rep. 2017, 7, 44350. [Google Scholar] [CrossRef]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G.; et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Cross, K.L.; Campbell, J.H.; Balachandran, M.; Campbell, A.G.; Cooper, C.J.; Griffen, A.; Heaton, M.; Joshi, S.; Klingeman, D.; Leys, E.; et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol. 2019, 37, 1314–1321. [Google Scholar] [CrossRef]
- Murugkar, P.P.; Collins, A.J.; Chen, T.; Dewhirst, F.E. Isolation and cultivation of candidate phyla radiation Saccharibacteria (TM7) bacteria in coculture with bacterial hosts. J. Oral Microbiol. 2020, 12, 1814666. [Google Scholar] [CrossRef]
- Bor, B.; Collins, A.J.; Murugkar, P.P.; Balasubramanian, S.; To, T.T.; Hendrickson, E.L.; Bedree, J.K.; Bidlack, F.B.; Johnston, C.D.; Shi, W.; et al. Insights obtained by culturing Saccharibacteria with their bacterial hosts. J. Dent. Res. 2020, 99, 685–694. [Google Scholar] [CrossRef]
- Ibrahim, A.; Maatouk, M.; Rajaonison, A.; Zgheib, R.; Haddad, G.; Bou Khalil, J.; Raoult, D.; Bittar, F. Adapted protocol for Saccharibacteria cocultivation: Two new members join the club of Candidate Phyla Radiation. Microbiol. Spectr. 2021, 9, e0106921. [Google Scholar] [CrossRef] [PubMed]
- Batinovic, S.; Rose, J.J.A.; Ratcliffe, J.; Seviour, R.J.; Petrovski, S. Cocultivation of an ultrasmall environmental parasitic bacterium with lytic ability against bacteria associated with wastewater foams. Nat. Microbiol. 2021, 6, 703–711. [Google Scholar] [CrossRef]
- Xie, B.; Wang, J.; Nie, Y.; Tian, J.; Wang, Z.; Chen, D.; Hu, B.; Wu, X.L.; Du, W. Type IV pili trigger episymbiotic association of Saccharibacteria with its bacterial host. Proc. Natl. Acad. Sci. USA 2022, 119, e2215990119. [Google Scholar] [CrossRef]
- Nie, J.; Utter, D.R.; Kerns, K.A.; Lamont, E.I.; Hendrickson, E.L.; Liu, J.; Wu, T.; He, X.; McLean, J.; Bor, B. Strain-level variation and diverse host bacterial responses in episymbiotic Saccharibacteria. mSystems 2022, 7, e0148821. [Google Scholar] [CrossRef]
- Yakimov, M.M.; Merkel, A.Y.; Gaisin, V.A.; Pilhofer, M.; Messina, E.; Hallsworth, J.E.; Klyukina, A.A.; Tikhonova, E.N.; Gorlenko, V.M. Cultivation of a vampire: Candidatus Absconditicoccus praedator. Environ. Microbiol. 2022, 24, 30–49. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Overholt, W.A.; Figueroa-Gonzalez, P.A.; Taubert, M.; Bornemann, T.L.V.; Probst, A.J.; Hölzer, M.; Marz, M.; Küsel, K. The economical lifestyle of CPR bacteria in groundwater allows little preference for environmental drivers. Environ. Microbiome 2021, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Gonzalez, P.A.; Bornemann, T.L.V.; Hinzke, T.; Maaß, S.; Trautwein-Schult, A.; Starke, J.; Moore, C.J.; Esser, S.P.; Plewka, J.; Hesse, T.; et al. Metaproteogenomics resolution of a high-CO2 aquifer community reveals a complex cellular adaptation of groundwater Gracilibacteria to a host-dependent lifestyle. Microbiome 2024, 12, 194. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, H.; Chen, Q.; Ni, J. Adaptation mechanism and ecological role of CPR bacteria in brackish-saline groundwater. NPJ Biofilms Microbiomes 2024, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Hu, Y.; Liu, L.; Liu, S.J.; Zhang, T. Genome-centric metagenomics reveals the host-driven dynamics and ecological role of CPR bacteria in an activated sludge system. Microbiome 2023, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Bor, B.; Bedree, J.K.; Shi, W.; McLean, J.S.; He, X. Saccharibacteria (TM7) in the human oral microbiome. J. Dent. Res. 2019, 98, 500–509. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Z.; Zhang, Y.; Wan, H.; Zhao, H. Tetrabromobisphenol A biotransformation in aged soil: Mechanism analysis induced by root exudates during rhizoremediation of Helianthus annus. J. Hazard Mater. 2024, 480, 136089. [Google Scholar] [CrossRef]
- Vinyes-Nadal, M.; Kümmel, S.; Espín, Y.; Gómez-Alday, J.J.; Gehre, M.; Otero, N.; Torrentó, C. Dual C and Cl compound-specific isotope analysis and metagenomic insights into the degradation of the pesticide methoxychlor. J. Hazard Mater. 2024, 480, 135929. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Järvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE 2012, 7, e31386. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Feng, K.; Peng, X.; Zhang, Z.; Gu, S.; He, Q.; Shen, W.; Wang, Z.; Wang, D.; Hu, Q.; Li, Y.; et al. iNAP: An integrated network analysis pipeline for microbiome studies. Imeta 2022, 1, e13. [Google Scholar] [CrossRef]
- Kauffman, J.; Kittas, A.; Bennett, L.; Tsoka, S. DyCoNet: A Gephi plugin for community detection in dynamic complex networks. PLoS ONE 2014, 9, e101357. [Google Scholar] [CrossRef]
- Man, D.K.W.; Hermans, S.M.; Taubert, M.; Garcia, S.L.; Hengoju, S.; Küsel, K.; Rosenbaum, M.A. Enrichment of different taxa of the enigmatic candidate phyla radiation bacteria using a novel picolitre droplet technique. ISME Commun. 2024, 4, ycae080. [Google Scholar] [CrossRef] [PubMed]
- Luef, B.; Frischkorn, K.R.; Wrighton, K.C.; Holman, H.Y.; Birarda, G.; Thomas, B.C.; Singh, A.; Williams, K.H.; Siegerist, C.E.; Tringe, S.G.; et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat. Commun. 2015, 6, 6372. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, J.; Yu, X.; Chen, G.; Yu, Z. Patterns in the composition of microbial communities from a subtropical river: Effects of environmental, spatial and temporal factors. PLoS ONE 2013, 8, e81232. [Google Scholar] [CrossRef]
- Kong, Z.; Kou, W.; Ma, Y.; Yu, H.; Ge, G.; Wu, L. Seasonal dynamics of the bacterioplankton community in a large, shallow, highly dynamic freshwater lake. Can. J. Microbiol. 2018, 64, 786–797. [Google Scholar] [CrossRef]
- Yan, L.; Hermans, S.M.; Totsche, K.U.; Lehmann, R.; Herrmann, M.; Küsel, K. Groundwater bacterial communities evolve over time in response to recharge. Water Res. 2021, 201, 117290. [Google Scholar] [CrossRef]
- Fierer, N.; Holland-Moritz, H.; Alexiev, A.; Batther, H.; Dragone, N.B.; Friar, L.; Gebert, M.J.; Gering, S.; Henley, J.B.; Jech, S.; et al. A Metagenomic Investigation of Spatial and Temporal Changes in Sewage Microbiomes across a University Campus. mSystems 2022, 7, e0065122. [Google Scholar] [CrossRef]
- Geesink, P.; Wegner, C.E.; Probst, A.J.; Herrmann, M.; Dam, H.T.; Kaster, A.K.; Küsel, K. Genome-inferred spatio-temporal resolution of an uncultivated Roizmanbacterium reveals its ecological preferences in groundwater. Environ. Microbiol. 2020, 22, 726–737. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Maatouk, M.; Ibrahim, A.; Rolain, J.M.; Merhej, V.; Bittar, F. Small and Equipped: The Rich Repertoire of Antibiotic Resistance Genes in Candidate Phyla Radiation Genomes. mSystems 2021, 6, e0089821. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Seong, H.J.; Johnson, T.A.; Cha, C.J.; Sul, W.J.; Chae, J.C. Persistence of antibiotic resistance from animal agricultural effluents to surface water revealed by genome-centric metagenomics. J. Hazard. Mater. 2023, 457, 131761. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Q.; Zhong, H.; Liu, S.; Sun, W. CPR bacteria and DPANN archaea play pivotal roles in response of microbial community to antibiotic stress in groundwater. Water Res. 2024, 251, 121137. [Google Scholar] [CrossRef]
- Wang, J.; Shan, S.; Li, D.; Zhang, Z.; Ma, Q. Long-term influence of chloroxylenol on anaerobic microbial community: Performance, microbial interaction, and antibiotic resistance gene behaviors. Sci. Total Environ. 2023, 897, 165330. [Google Scholar] [CrossRef] [PubMed]
- Xin, R.; Li, K.; Ding, Y.; Zhang, K.; Qin, M.; Jia, X.; Fan, P.; Li, R.; Zhang, K.; Yang, F. Tracking the extracellular and intracellular antibiotic resistance genes across whole year in wastewater of intensive dairy farm. Ecotoxicol. Environ. Saf. 2024, 269, 115773. [Google Scholar] [CrossRef]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network analysis methods for studying microbial communities: A mini review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Faust, K. Open challenges for microbial network construction and analysis. ISME J. 2021, 15, 3111–3118. [Google Scholar] [CrossRef]
- Zamkovaya, T.; Foster, J.S.; de Crécy-Lagard, V.; Conesa, A. A network approach to elucidate and prioritize microbial dark matter in microbial communities. ISME J. 2021, 15, 228–244. [Google Scholar] [CrossRef]
- Srinivasan, S.; Jnana, A.; Murali, T.S. Modeling Microbial Community Networks: Methods and Tools for Studying Microbial Interactions. Microb. Ecol. 2024, 87, 56. [Google Scholar] [CrossRef]
- Chiriac, M.C.; Bulzu, P.A.; Andrei, A.S.; Okazaki, Y.; Nakano, S.I.; Haber, M.; Kavagutti, V.S.; Layoun, P.; Ghai, R.; Salcher, M.M. Ecogenomics sheds light on diverse lifestyle strategies in freshwater CPR. Microbiome 2022, 10, 84. [Google Scholar] [CrossRef]
- Zhao, R.; Farag, I.F.; Jørgensen, S.L.; Biddle, J.F. Occurrence, Diversity, and Genomes of “Candidatus Patescibacteria” along the Early Diagenesis of Marine Sediments. Appl. Environ. Microbiol. 2022, 88, e0140922. [Google Scholar] [CrossRef] [PubMed]
- Momper, L.; Casar, C.P.; Osburn, M.R. A metagenomic view of novel microbial and metabolic diversity found within the deep terrestrial biosphere at DeMMO: A microbial observatory in South Dakota, USA. Environ. Microbiol. 2023, 25, 3719–3737. [Google Scholar] [CrossRef]
- Santana-Pereira, A.L.R.; Moen, F.S.; Severance, B.; Liles, M.R. Influence of soil nutrients on the presence and distribution of CPR bacteria in a long-term crop rotation experiment. Front. Microbiol. 2023, 14, 1114548. [Google Scholar] [CrossRef]
- Mukhia, S.; Kumar, A.; Kumar, R. Bacterial community distribution and functional potentials provide key insights into their role in the ecosystem functioning of a retreating Eastern Himalayan glacier. FEMS Microbiol. Ecol. 2024, 100, fiae012. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Chain, P.S.; Grafham, D.V.; Fulton, R.S.; Fitzgerald, M.G.; Hostetler, J.; Muzny, D.; Ali, J.; Birren, B.; Bruce, D.C.; Buhay, C.; et al. Genomics. Genome project standards in a new era of sequencing. Science 2009, 326, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Méheust, R.; Burstein, D.; Castelle, C.J.; Banfield, J.F. The distinction of CPR bacteria from other bacteria based on protein family content. Nat. Commun. 2019, 10, 4173. [Google Scholar] [CrossRef]
- Bokhari, R.H.; Amirjan, N.; Jeong, H.; Kim, K.M.; Caetano-Anollés, G.; Nasir, A. Bacterial Origin and Reductive Evolution of the CPR Group. Genome Biol. Evol. 2020, 12, 103–121. [Google Scholar] [CrossRef]
- Probst, A.J.; Elling, F.J.; Castelle, C.J.; Zhu, Q.; Elvert, M.; Birarda, G.; Holman, H.N.; Lane, K.R.; Ladd, B.; Ryan, M.C.; et al. Lipid analysis of CO2-rich subsurface aquifers suggests an autotrophy-based deep biosphere with lysolipids enriched in CPR bacteria. ISME J. 2020, 14, 1547–1560. [Google Scholar] [CrossRef]
- Probst, A.J.; Castelle, C.J.; Singh, A.; Brown, C.T.; Anantharaman, K.; Sharon, I.; Hug, L.A.; Burstein, D.; Emerson, J.B.; Thomas, B.C.; et al. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ. Microbiol. 2017, 19, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.V.; Akita, H.; Strott, C.A. Molecular cloning, expression, and characterization of human bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase and its functional domains. J. Biol. Chem. 1998, 273, 19311–19320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.B.; Li, Y.Q.; Lin, J.Q.; Pang, X.; Liu, X.M.; Liu, B.Q.; Wang, R.; Zhang, C.J.; Wu, Y.; Lin, J.Q.; et al. The two-component system RsrS-RsrR regulates the tetrathionate intermediate pathway for thiosulfate oxidation in Acidithiobacillus caldus. Front. Microbiol. 2016, 7, 1755. [Google Scholar] [CrossRef] [PubMed]
- Kappler, U.; Bennett, B.; Rethmeier, J.; Schwarz, G.; Deutzmann, R.; McEwan, A.G.; Dahl, C. Sulfite: Cytochrome c oxidoreductase from Thiobacillus novellus. purification, characterization, and molecular biology of a heterodimeric member of the sulfite oxidase family. J. Biol. Chem. 2000, 275, 13202–13212. [Google Scholar] [CrossRef]
- Beam, J.P.; Becraft, E.D.; Brown, J.M.; Schulz, F.; Jarett, J.K.; Bezuidt, O.; Poulton, N.J.; Clark, K.; Dunfield, P.F.; Ravin, N.V.; et al. Ancestral absence of electron transport chains in Patescibacteria and DPANN. Front. Microbiol. 2020, 11, 1848. [Google Scholar] [CrossRef]
- Borisov, V.B.; Nastasi, M.R.; Forte, E. Cytochrome bd as antioxidant redox enzyme. Mol. Biol. 2023, 57, 1077–1084. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, K.; Ye, L.; Cao, C.; Che, G.; Wang, Y.; Hong, Y. Multi-Metagenome Analysis Unravels Community Collapse After Sampling and Hints the Cultivation Strategy of CPR Bacteria in Groundwater. Microorganisms 2025, 13, 972. https://doi.org/10.3390/microorganisms13050972
Jiang K, Ye L, Cao C, Che G, Wang Y, Hong Y. Multi-Metagenome Analysis Unravels Community Collapse After Sampling and Hints the Cultivation Strategy of CPR Bacteria in Groundwater. Microorganisms. 2025; 13(5):972. https://doi.org/10.3390/microorganisms13050972
Chicago/Turabian StyleJiang, Kai, Lijia Ye, Chunling Cao, Gen Che, Yanxing Wang, and Yu Hong. 2025. "Multi-Metagenome Analysis Unravels Community Collapse After Sampling and Hints the Cultivation Strategy of CPR Bacteria in Groundwater" Microorganisms 13, no. 5: 972. https://doi.org/10.3390/microorganisms13050972
APA StyleJiang, K., Ye, L., Cao, C., Che, G., Wang, Y., & Hong, Y. (2025). Multi-Metagenome Analysis Unravels Community Collapse After Sampling and Hints the Cultivation Strategy of CPR Bacteria in Groundwater. Microorganisms, 13(5), 972. https://doi.org/10.3390/microorganisms13050972