Rhizosphere Metagenomics of Paspalum scrobiculatum L. (Kodo Millet) Reveals Rhizobiome Multifunctionalities

 ,
,  ,
,  and
and
Abstract
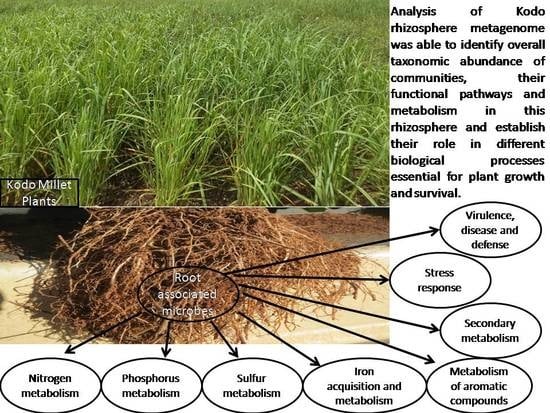
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Analysis
2.2. Metagenomic DNA Extraction and Sequencing
2.3. Annotation of Metagenomic Dataset
2.4. Taxonomic and Functional Annotation
2.5. Metabolic Potential Analysis
2.6. Availability of Data and Associated Information
3. Results and Discussion
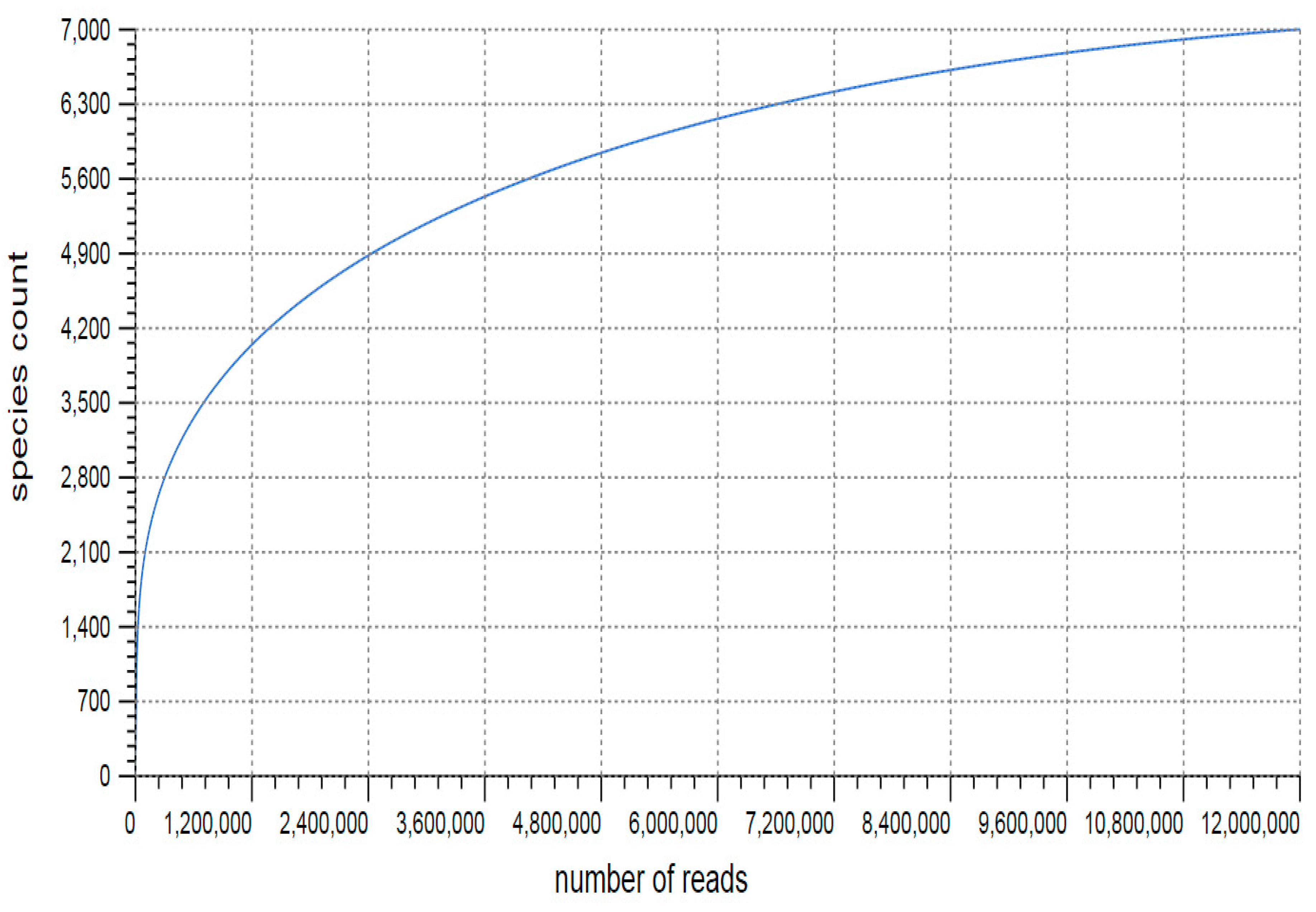
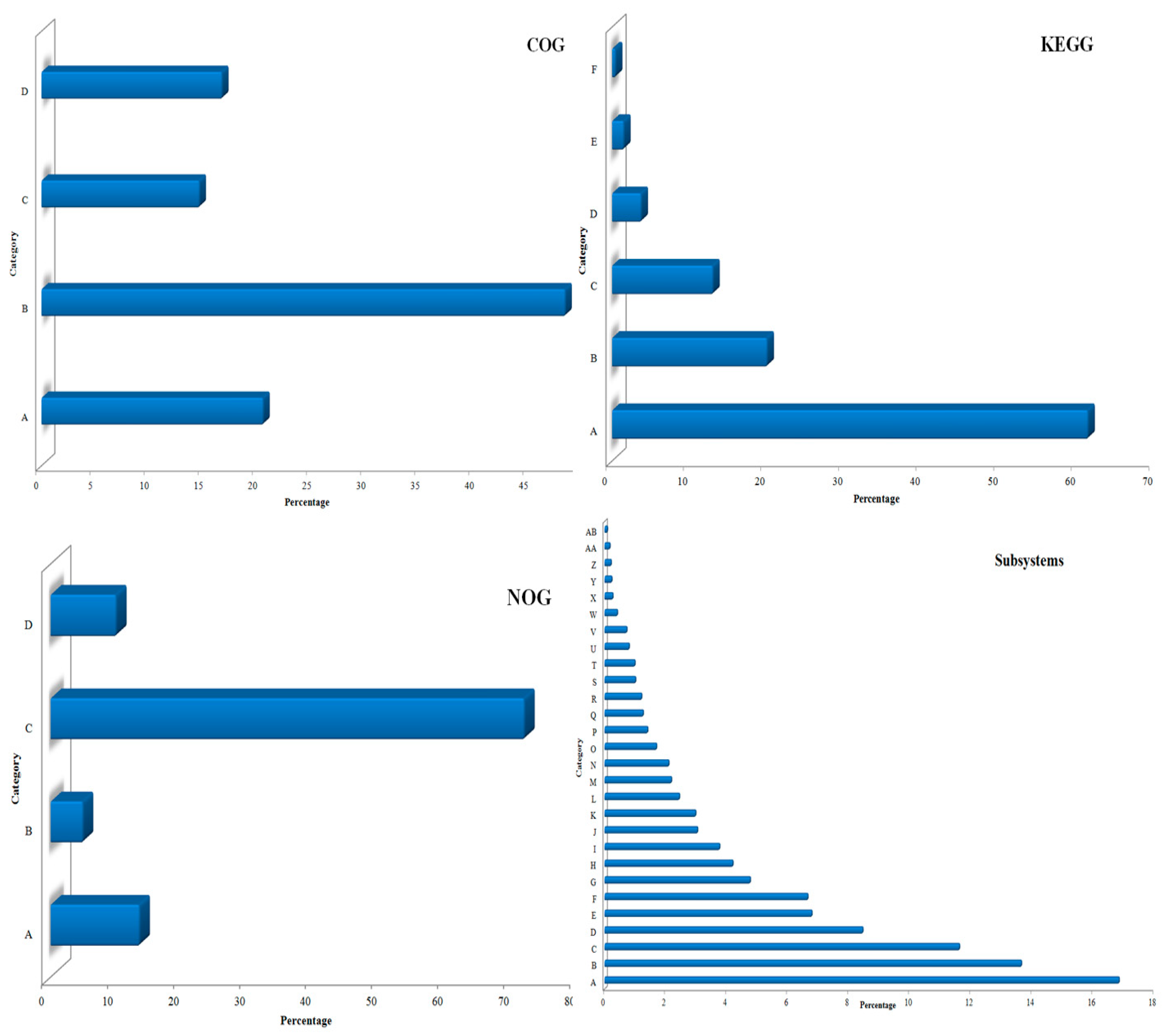
3.1. Sequencing and Annotation of Proteins
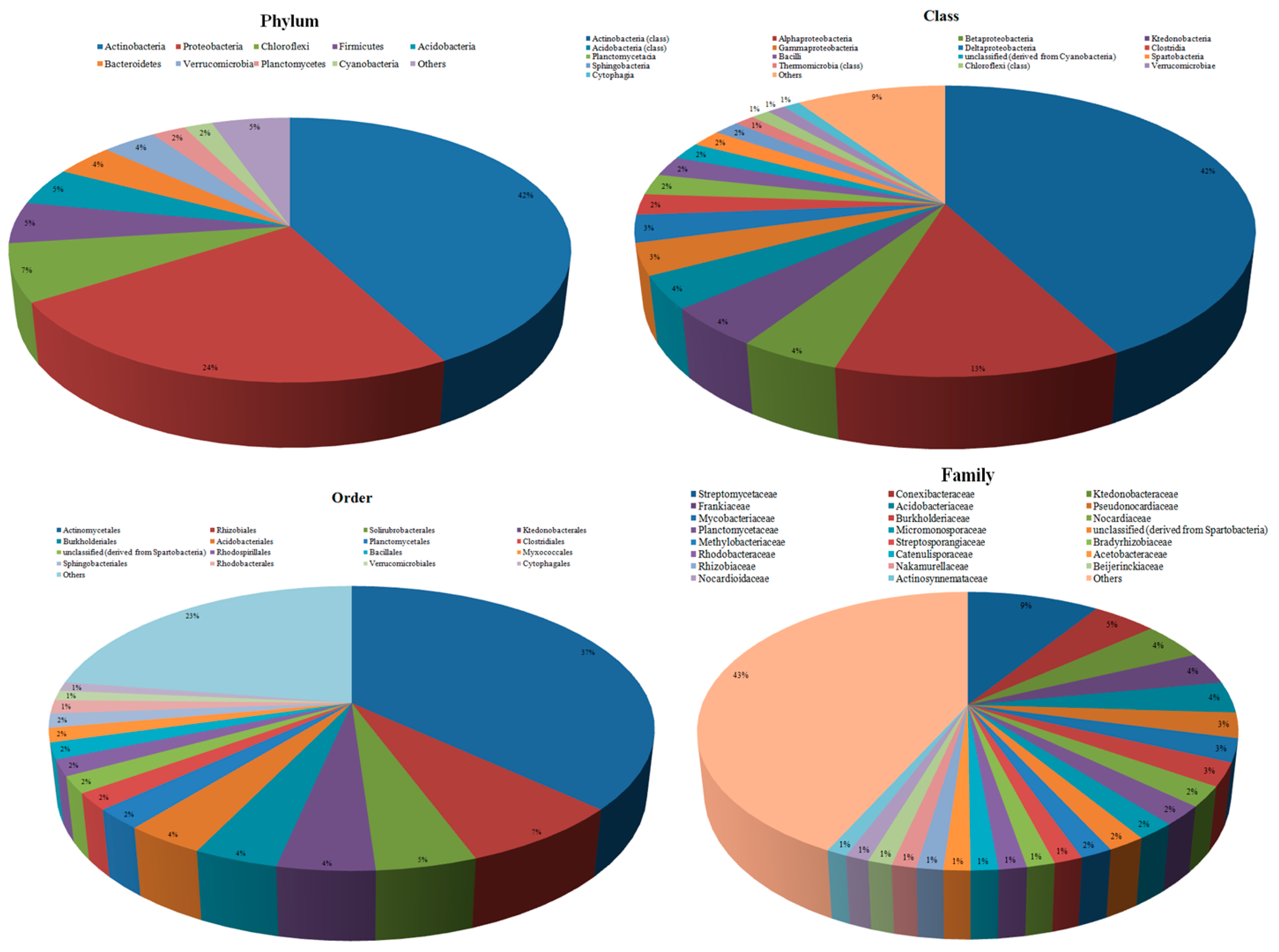
3.2. Taxonomic Microbial Diversity in the Kodo Rhizosphere
3.3. Community Composition and Abundance
3.4. Metabolic Multifunctionalities in the Kodo Rhizosphere
3.5. Carbon Fixation
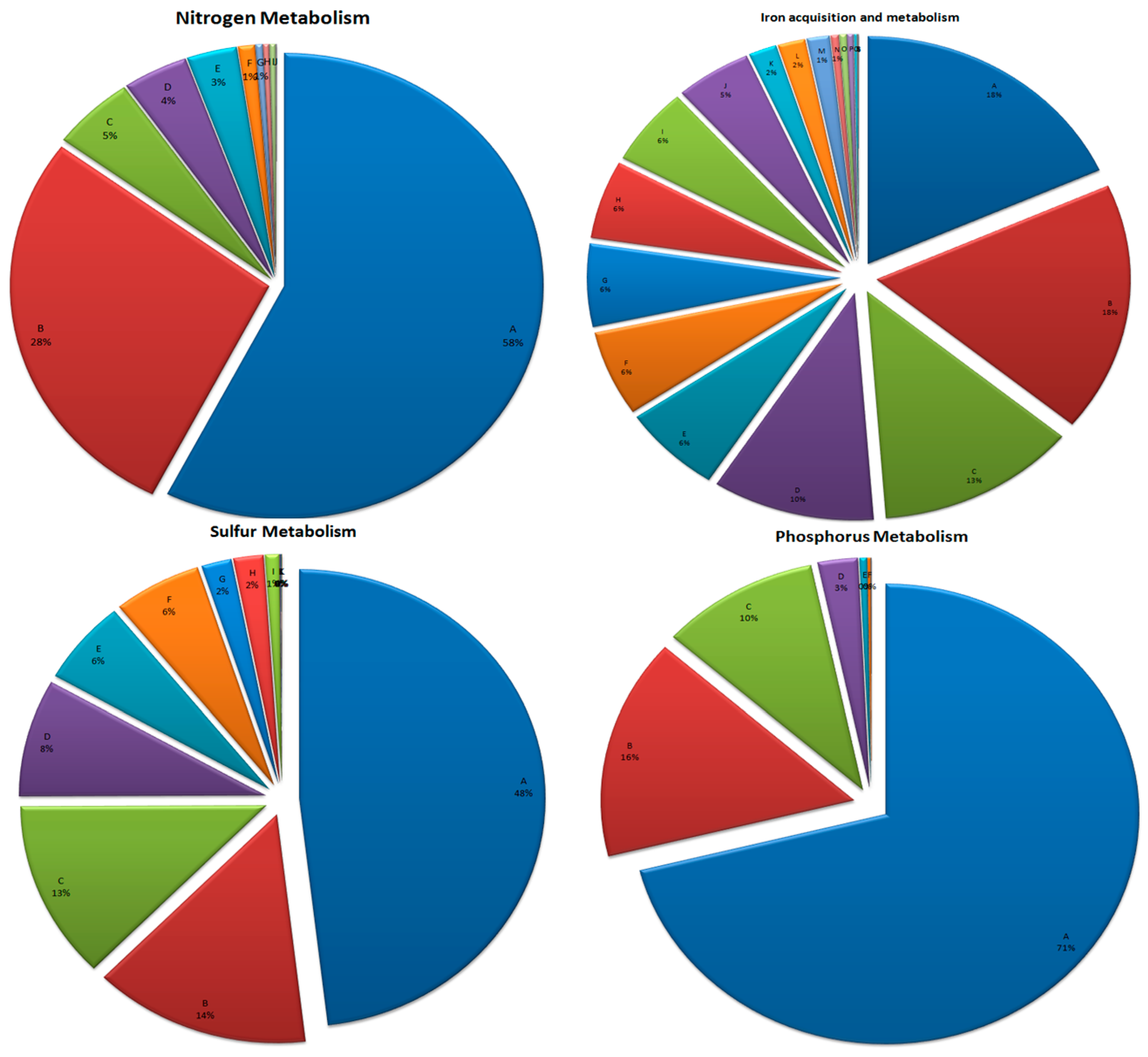
3.6. Mineral Metabolism
3.6.1. Nitrogen Metabolism
3.6.2. Phosphorus Metabolism
3.6.3. Sulfur Metabolism
3.6.4. Iron Acquisition and Metabolism
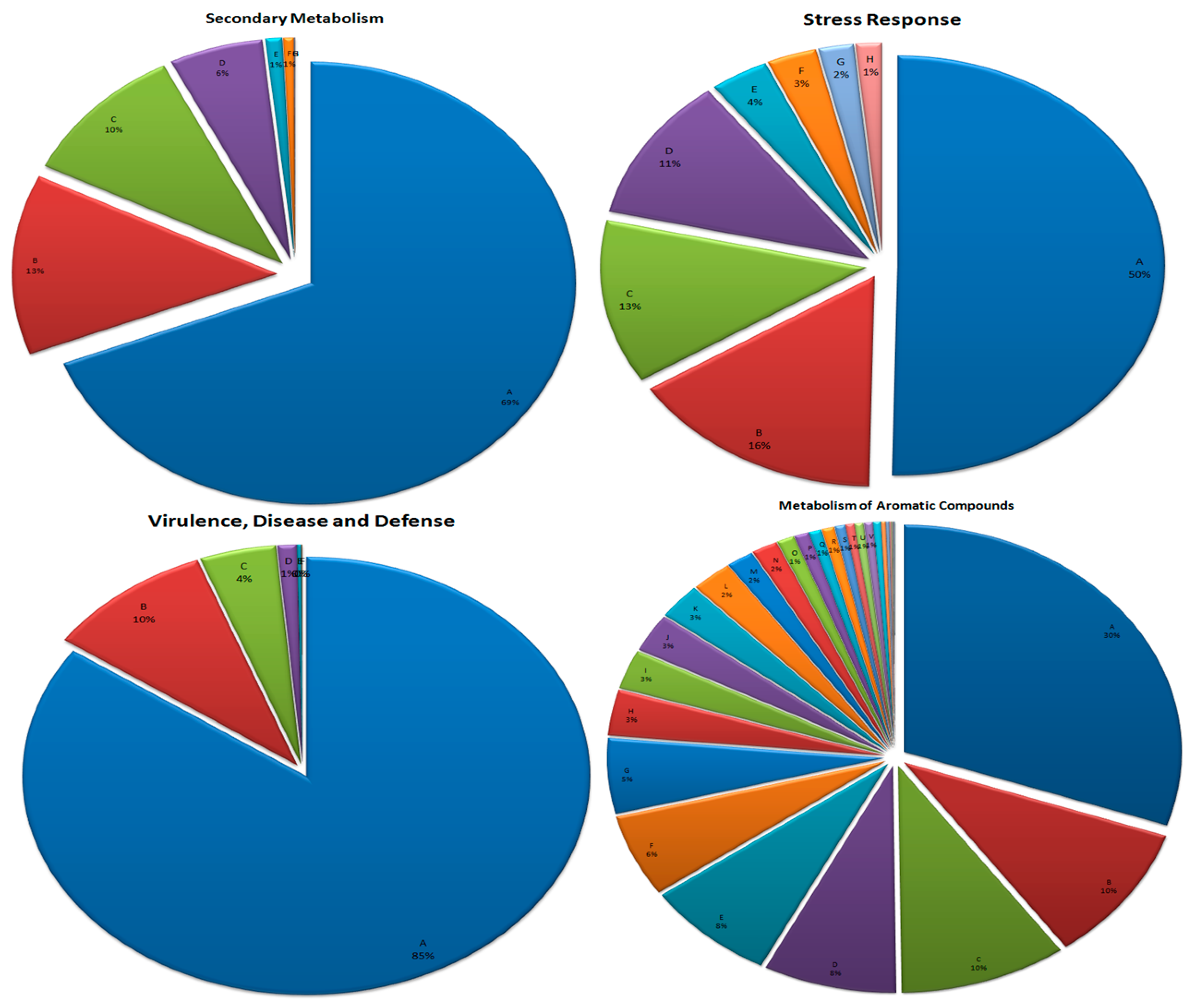
3.7. Metabolism of Aromatic Compounds
3.8. Secondary Metabolism
3.9. Stress Response
3.10. Virulence, Disease, and Defense
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lareen, A.; Burton, F.; Schafer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef]
- Xu, Y. Envirotyping for deciphering environmental impacts on crop plants. Theor. Appl. Genet. 2016, 129, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing unculturable bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Gunderman, L.; Coles, C.A.; Lochmann, J.; Parks, M.; Ballard, E. 16S rRNA Gene-based metagenomic analysis of ozark cave bacteria. Diversity 2017, 9, 31. [Google Scholar] [CrossRef]
- Rosselló-Mora, R.; Amann, R. The species concept for prokaryotes. FEMS Microbiol. Rev. 2001, 25, 39–67. [Google Scholar] [CrossRef]
- Torsvik, V.; Qvreas, L. Microbial diversity and function in soil: From genes to ecosystem. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Biotechnological prospects from metagenomics. Curr. Opin. Microbiol. 2003, 14, 303–310. [Google Scholar] [CrossRef]
- Parnell, J.J.; Berka, R.; Young, H.A.; Sturino, J.M.; Kang, Y.; Barnhart, D.M.; DiLeo, M.V. From the Lab to the Farm: An Industrial Perspective of Plant Beneficial Microorganisms. Front. Plant Sci. 2016, 7, 1110. [Google Scholar] [CrossRef]
- de Wet, J.M.J.; Rao, K.E.P.; Mengesha, M.H.; Brink, D.E. Diversity in kodo millet, Paspalum scrobiculatum. Econ. Bot. 1983, 37, 159–163. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chandra, T.S. ESR spectroscopic study reveals higher free radical quenching potential in kodo millet (Paspalum scrobiculatum) compared to other millets. Food Chem. 2005, 92, 177–182. [Google Scholar] [CrossRef]
- Yadav, N.; Chaudhary, K.; Singh, A.; Gupta, A. Evaluation of hypoglycemic properties of kodo millet based food products in healthy subjects. IOSR J. Pharm. 2013, 3, 14–20. [Google Scholar]
- Deshpande, S.S.; Mohapatra, D.; Tripathi, M.K.; Sadvatha, R.H. Kodo millet-nutritional value and utilization in Indian foods. J. Grain Proc. Storage 2015, 2, 16–23. [Google Scholar]
- Alsterberg, C.; Roger, F.; Sundbäck, K.; Juhanson, J.; Hulth, S.; Hallin, S.; Gamfeldt, L. Diversity of habitats and bacterial communities support landscape-scale multifunctionality differently across seasons. Peer J. 2016, 4, e2036v1. [Google Scholar] [CrossRef]
- Prabhakar; Prabhu, C.G.; Boraiah, B.; Bhat, S.; Nandani, C.; Kiran, T.V.; Manjunath, H.A. Improved Production Technology for Kodo Millet; Technical Bulletin-2/2017-18; Project Coordinating Unit, ICAR-AICRP on Small Millets; GKVK: Bengaluru, India, 2017. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinf. 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2015, 43, D30–D35. [Google Scholar] [CrossRef]
- Chen, B.; Yang, Y.; Liang, X.; Yu, K.; Zhang, T.; Li, X. Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Imchen, M.; Kumavath, R.; Barh, D.; Azevedo, V.; Ghosh, P.; Viana, M.; Wattam, A.R. Searching for signatures across microbial communities: Metagenomic analysis of soil samples from mangrove and other ecosystems. Sci. Rep. 2017, 7, 8859. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, S.; Schumann, P. Introduction to the taxonomy of actinobacteria. Prokaryotes 2006, 3, 297–321. [Google Scholar]
- Sellstedt, A.; Richau, K.H. Aspects of nitrogen-fixing Actinobacteria, in particular free-living and symbiotic Frankia. FEMS Microbiol. Lett. 2013, 342, 179–186. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D. Complete genome sequence of the model actinomycete Streptomyces Coelicolor A3 (2). Nature 2002, 417, 141. [Google Scholar] [CrossRef]
- Waksman, S.A.; Schatz, A.; Reynolds, D.M. Production of antibiotic substances by actinomycetes. Ann. NY Acad. Sci. 2010, 48, 73–86. [Google Scholar] [CrossRef]
- Bérdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385. [Google Scholar] [CrossRef]
- Daoudi-Hamdad, H.H.; Bhatnagar, F.; Baratti, T.; Lefebvre, G. H107, a new aminoglycoside anti-Pseudomonas antibiotic produced by a new strain of Spirillospora. Microbios 2000, 102, 69–77. [Google Scholar]
- Bibb, M.J. Regulation of secondary metabolism in Streptomyces. Curr. Opin. Microbiol. 2005, 8, 208–215. [Google Scholar] [CrossRef]
- Forar, L.R.; Amany, K.; Ali, E.; Arab, B.C. Taxonomy, Identification and biological activities of a novel isolate of Streptomyces tendae. J. Biotechnol. 2006, 9, 427–436. [Google Scholar]
- Janardhan, A.; Kumar, A.P.; Viswanath, B.; Saigopal, D.V.R.; Narasimha, G. Production of bioactive compounds by actinomycetes and their antioxidant properties. Biotechnol. Res. Int. 2014, 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Schweder, T.; Lindequist, U.; Lalk, M. Screening for new metabolites from marine microorganisms. In Marine Biotechnology I. Advances in Biochemical Engineering/Biotechnology; Ulber, R., Le Gal, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 96, pp. 1–48. [Google Scholar]
- Tjepkema, J.D.; Cashon, R.E.; Beckwith, J.; Schwintzer, C.R. Hemoglobin in Frankia, a nitrogen-fixing actinomycete. Appl. Environ. Microbiol. 2002, 68, 2629–2631. [Google Scholar] [CrossRef] [PubMed]
- Zakhia, F.; Jeder, H.; Willems, A.; Gillis, M.; Dreyfus, B.; de Lajudie, P. Diverse bacteria associated with root nodules of spontaneous legumes in Tunisia and first report for nifH-like gene within the genera Microbacterium and Starkeya. Microb. Ecol. 2006, 51, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Gtari, M.; Ghodhbane-Gtari, F.; Nouioui, I.; Beauchemin, N.; Tisa, L.S. Phylogenetic perspectives of nitrogen-fixing actinobacteria. Arch. Microbiol. 2012, 194, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liang, J.; Liu, R.; Hu, C.; Qu, J. Metagenomic analysis reveals microbial diversity and function in the rhizosphere soil of a constructed wetland. Environ. Technol. 2014, 35, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Moat, A.G.; Foster, J.W.; Spector, M.P. Microbial Physiology; John Wiley & Sons: Hoboken, NJ, USA, 2002; ISBN 0-471-39483-1. [Google Scholar]
- Joice, R.; Yasuda, K.; Shafquat, A.; Morgan, X.C.; Huttenhower, C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab. 2014, 20, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.J.; Verberkmoes, N.C.; Thelen, M.P.; Tyson, G.W.; Baker, B.J.; Blake, R.C.; Shah, M., 2nd; Hettich, R.L.; Banfield, J.F. Community proteomics of a natural microbial biofilm. Science 2005, 308, 1915–1920. [Google Scholar] [CrossRef]
- Bertin, P.N.; Heinrich-Salmeron, A.; Pelletier, E.; Goulhen-Chollet, F.; Arsène-Ploetze, F.; Gallien, S. Metabolic diversity among main microorganisms inside an arsenic-rich ecosystem revealed by meta-and proteo-genomics. ISMEJ 2011, 5, 1735. [Google Scholar] [CrossRef]
- Parro, V.; Moreno-Paz, M.; Gonzalez-Toril, E. Analysis of environmental transcriptomes by DNA microarrays. Environ. Microbiol. 2007, 9, 453–464. [Google Scholar] [CrossRef]
- Moreno-Paz, M.; Go´mez, M.J.; Arcas, A.; Parro, V. Environmental transcriptome analysis reveals physiological differences between biofilm and planktonic modes of life of the iron oxidizing bacteria Leptospirillum spp. in their natural microbial community. BMC Genom. 2010, 11, 404. [Google Scholar] [CrossRef]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Urich, T. Simultaneous assessment of soil microbial community structure and function htrough analysis of the meta-transcriptome. PLoS ONE 2008, 3, e2527. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.A. Metatranscriptomics: Eavesdropping on complex microbial communities. Microbe 2009, 4, 329–334. [Google Scholar] [CrossRef][Green Version]
- Chen, L.X. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 2015, 9, 1579. [Google Scholar] [CrossRef] [PubMed]
- Masclaux-Daubresse, C. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, B.S.; Rawat, A.K.; Dixit, B.K.; Tahkur, R.K. Effect of inputs integration on yield, uptake and economics of Kodo Millet (Paspalum scrobiculatum L.). Econ. Aff. 2016, 61, 519–524. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Fernández-González, A.J.; Villadas, P.J.; Robles, A.B.; Toro, N.; Fernández-López, M. Metagenomic assessment of the potential microbial nitrogen pathways in the rhizosphere of a mediterranean forest after a wildfire. Microb. Ecol. 2015, 69, 895–904. [Google Scholar] [CrossRef]
- Minami, T.; Anda, M.; Mitsui, H.; Sugawara, M.; Kaneko, T.; Sato, S.; Minamisawa, K. Metagenomic analysis revealed methylamine and ureide utilization of soybean-associated Methylobacterium. Microb. Environ. 2016, 31, 268–278. [Google Scholar] [CrossRef]
- Qian, M.; Eaton, J.W.; Wolff, S. Cyanate-mediated inhibition of neutrophil myeloperoxidase activity. Biochem. J. 1997, 326, 159–166. [Google Scholar] [CrossRef]
- Purcarea, C. Aquifex aeolicus aspartate transcarbamoylase, an enzyme specialized for the efficient utilization of unstable carbamoyl phosphate at elevated temperature. J. Biol. Chem. 2003, 278, 52924–52934. [Google Scholar] [CrossRef]
- Ubalua, A.O. Cyanogenic glycosides and the fate of cyanide in soil. Aust. J. Crop. Sci. 2010, 4, 223–237. [Google Scholar]
- Widner, B.; Mulholland, M.R.; Mopper, K. Chromatographic determination of nanomolar cyanate concentrations in estuarine and sea waters by precolumn fluorescence derivatization. Anal. Chem. 2013, 85, 6661–6666. [Google Scholar] [CrossRef] [PubMed]
- Kamennaya, N.A.; Post, A.F. Distribution and expression of the cyanate acquisition potential among cyanobacterial populations in oligotrophic marine waters. Limnol. Oceanog. 2013, 58, 1959–1971. [Google Scholar] [CrossRef]
- Rocap, G. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 2003, 424, 1042. [Google Scholar] [CrossRef]
- Hatzenpichler, R. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc. Natl. Acad. Sci. USA 2008, 105, 2134–2139. [Google Scholar] [CrossRef]
- Palatinszky, M. Cyanate as an energy source for nitrifiers. Nature 2015, 524, 7563. [Google Scholar] [CrossRef]
- Philippot, L.; Hallin, S.; Schloter, M. Ecology of denitrifying prokaryotes in agricultural soil. Adv. Agron. 2007, 96, 249–305. [Google Scholar]
- Lamarche, M.G.; Wanner, B.L.; Creplin, S.; Harel, J. The phosphate regulon and bacterial virulence: A regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 2008, 32, 461–473. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Wright, M.S.; Stepanauskas, R.; McArthur, J.V. Co-selection of antibiotic and metal resistance. Trends Microbiol. 2006, 14, 176–182. [Google Scholar] [CrossRef]
- Sosa, O.A. Phosphorus redox reactions as pinch hitters in microbial metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 7–8. [Google Scholar] [CrossRef]
- Marschner, P.; Crowley, D.; Rengel, Z. Rhizosphere interactions between microorganisms and plants govern iron and phosphorus acquisition along the root axis-model and research methods. Soil. Biol. Biochem. 2011, 43, 883–894. [Google Scholar] [CrossRef]
- Ghosh, W.; Dam, B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 2009, 33, 999–1043. [Google Scholar] [CrossRef]
- Yousuf, B.; Kumar, R.; Mishra, A.; Jha, B. Unravelling the carbon and sulphur metabolism in coastal soil ecosystems using comparative cultivation-independent genome-level characterisation of microbial communities. PLoS ONE 2014, 9, e107025. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Stams, A.J. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Headd, B.; Engel, A.S. Evidence for niche partitioning revealed by the distribution of sulfur oxidation genes collected from areas of a terrestrial sulfidic spring with differing geochemical conditions. Appl. Environ. Microbiol. 2012, 79, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Tourova, T.P. Analysis of community composition of sulfur-oxidizing bacteria in hypersaline and soda lakes using sox B as a functional molecular marker. FEMS Microbiol. Ecol. 2013, 84, 280–289. [Google Scholar] [CrossRef]
- Yin, H. Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans. BMC Microbiol. 2014, 14, 179. [Google Scholar] [CrossRef]
- Zhang, X.; Niu, J.; Liang, Y.; Liu, X.; Yin, H. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 2016, 17, 21. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Carbon, iron and sulfur metabolism in acidophilic micro-oranisms. Adv. Microb. Physiol. 2009, 54, 201–255. [Google Scholar]
- Dopson, M.; Johnson, D.B. Biodiversity, metabolism and applications of acidophilic sulfur-metabolizing microorganisms. Environ. Microbiol. 2012, 14, 2620–2631. [Google Scholar] [CrossRef]
- Rout, G.R.; Sahoo, S. Role of iron in plant growth and metabolism. Rev. Agric. Sci. 2015, 3, 1–24. [Google Scholar] [CrossRef]
- Menhart, N.; Thariath, A.; Viswanatha, T. Characterization of the pyoverdines of Azotobacter vinelandii ATCC12837 with regard to heterogeneity. Biol. Met. 1991, 4, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Berti, A.D.; Thomas, M.G. Analysis of achromobactin biosynthesis by Pseudomonas syringae pv. syringae B728a. J. Bacteriol. 2009, 191, 4594–4604. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.D.; Balbo, P.B.; Jones, H.A.; Fetherston, J.D.; DeMoll, E. Yersiniabactin from Yersinia pestis: Biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology 1999, 145, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kim, C.Y.; Fox, D.T.; Koppisch, A.T. Siderophore-mediated iron acquisition in Bacillus anthracis and related strains. Microbiology 2010, 156, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Brandel, J.; Humbert, N.; Elhabiri, M.; Schalk, I.J.; Mislin, G.L.; Albrecht-Gary, A.M. Pyochelin, a siderophore of Pseudomonas aeruginosa: Physicochemical characterization of the iron (III), copper (II) and zinc (II) complexes. Dalton Trans. 2012, 41, 2820–2834. [Google Scholar] [CrossRef]
- Das, S.; Bora, S.S.; Yadav, R.N.S.; Barooah, M. A metagenomic approach to decipher the indigenous microbial communities of arsenic contaminated groundwater of Assam. Genom. Data 2017, 12, 89–96. [Google Scholar] [CrossRef]
- Uchiyama, T.; Miyazaki, K. Metagenomic screening for aromatic compound-responsive transcriptional regulators. PLoS ONE 2013, 8, e75795. [Google Scholar] [CrossRef]
- Sun, W.; Krumins, V.; Fennell, D.E.; Kerkhof, L.J.; Häggblom, M.M. Anaerobic degradation of aromatic compounds. In Manual of Environmental Microbiology, Fourth Edition American Society of Microbiology; ASM Press: Washington, DC, USA, 2016; p. 5. [Google Scholar]
- Egland, P.G.; Harwood, C.S. BadR, a new MarR family member, regulates anaerobic benzoate degradation by Rhodopseudomonas palustris in concert with AadR, an Fnr family member. J. Bacteriol. 1999, 181, 2102–2109. [Google Scholar]
- Koutros, S. Heterocyclic aromatic amine pesticide use and human cancer risk: Results from the US Agricultural Health Study. Int. J. Cancer 2009, 124, 1206–1212. [Google Scholar] [CrossRef]
- Grund, E.; Denecke, B.; Eichenlaub, R. Naphthalene degradation via salicylate and gentisate by Rhodococcus sp. strain B4. Appl. Environ. Microbiol. 1992, 58, 1874–1877. [Google Scholar] [PubMed]
- Liu, T.T. Functional characterization of a gene cluster involved in gentisate catabolism in Rhodococcus sp. strain NCIMB 12038. Appl. Microbiol. Biotechnol. 2011, 90, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.S.; Qian, K.; Robbins, W.K. Nitrogen speciation of polar petroleum compounds by compound class separation and on-line liquid chromatography–mass spectrometry (LC-MS). J. Sep. Sci. 1994, 17, 271–276. [Google Scholar] [CrossRef]
- Yang, M. Isolation and identification of a carbazole degradation gene cluster from Sphingomonas sp. JS1. World J. Microbiol. Biotechnol. 2009, 25, 1625. [Google Scholar] [CrossRef]
- Teufel, R. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. USA 2010, 107, 14390–14395. [Google Scholar] [CrossRef]
- Walsh, C.T.; Fischbach, M.A. Natural products version 2.0: Connecting genes to molecules. J. Am. Chem. Soc. 2010, 132, 2469–2493. [Google Scholar] [CrossRef]
- Banik, J.J.; Brady, S.F. Recent application of metagenomic approaches toward the discovery of antimicrobials and other bioactive small molecules. Curr. Opin. Microbiol. 2010, 13, 603–609. [Google Scholar] [CrossRef]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Wilson, M.R.; Zha, L.; Balskus, E.P. Natural product discovery from the human microbiome. J. Biol. Chem. 2017, 292, 8546–8552. [Google Scholar] [CrossRef]
- Cimermancic, P. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 7, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escribano, J.; Bibb, M. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Kim, H.; Charusanti, P.; Palsson, B.; Lee, S. Systems biology and biotechnology ofStreptomyces species for the production of secondary metabolites. Biotechnol. Adv. 2014, 32, 255–268. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.E. Engineering of secondary metabolism. Annu. Rev. Genet. 2015, 49, 71–94. [Google Scholar] [CrossRef]
- Müller, C.A.; Obermeier, M.M.; Berg, G. Bioprospecting plant-associated microbiomes. J. Biotechnol. 2016, 235, 171–180. [Google Scholar] [CrossRef]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes–a review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef]
- Gunatilaka, A.A.L. Natural products from plant-associated microorganisms: Distribution, structural diversity, bioactivity, and implications of their occurrence. J. Nat. Prod. 2006, 69, 509–526. [Google Scholar] [CrossRef]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Salvador, V.H. Cinnamic acid increases lignin production and inhibits soybean root growth. PLoS ONE 2013, 8, e69105. [Google Scholar] [CrossRef]
- Chavan, S. Reduced glutathione: Importance of specimen collection. Ind. J. Clin. Biochem. 2005, 20, 150–152. [Google Scholar] [CrossRef]
- Cascella, R. S-linolenoyl glutathione intake extends life-span and stress resistance via Sir-2.1 upregulation in Caenorhabditis elegans. Free Radic. Biol. Med. 2014, 73, 127–135. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L. Nuclear glutathione. Biochim. Biophys. Acta 2013, 1830, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Pallardó, F.V. Maintenance of glutathione levels and its importance in epigenetic regulation. Front. Pharmacol. 2014, 5, 88. [Google Scholar]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Alonso, A.; Sanchez, P.; Martinez, J.L. Environmental selection of antibiotic resistance genes. Environ. Microbiol. 2001, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.V.; Vyas, B.R.M. Screening and characterization of plant growth and health promoting rhizobacteria. Int. J. Curr. Microbiol. Appl. Sci. 2014, 3, 139–155. [Google Scholar]
- Coutte, L. Role of adhesin release for mucosal colonization by a bacterial pathogen. J. Exp. Med. 2003, 197, 735–742. [Google Scholar] [CrossRef]
- Shi, P. Metagenomic insights into chlorination effects on microbial antibiotic resistance in drinking water. Water Res. 2013, 47, 111–120. [Google Scholar] [CrossRef]
- Kristiansson, E. Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS ONE 2011, 6, e17038. [Google Scholar] [CrossRef]
- Monier, J.M. Metagenomic exploration of antibiotic resistance in soil. Curr. Opin. Microbiol. 2011, 14, 229–235. [Google Scholar] [CrossRef]
- Medeiros, J.D. Comparative metagenome of a stream impacted by the urbanization phenomenon. Braz. J. Microbiol. 2016, 47, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Metagenomic profiling of antibiotic resistance genes and mobile genetic elements in a tannery wastewater treatment plant. PLoS ONE 2013, 8, e76079. [Google Scholar] [CrossRef] [PubMed]
- Hamonts, K. Field study reveals core plant microbiota and relative importance of their drivers. Environ. Microbiol. 2018, 20, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moënne-Loccoz, Y. Let the core microbiota be functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D.; et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 4894. [Google Scholar] [CrossRef]
- Jin, T. Taxonomic structure and functional association of foxtail millet root microbiome. Gigascience 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef]
- Trivedi, P. Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J. 2012, 6, 363–383. [Google Scholar] [CrossRef]
- Riera, N.; Handique, U.; Zhang, Y.; Dewdney, M.M.; Wang, N. Characterization of antimicrobial-producing beneficial bacteria isolated from Huanglongbing Escape citrus trees. Front. Microbiol. 2017, 8, 2415. [Google Scholar] [CrossRef]
- Prabha, R.; Singh, D.P.; Verma, M.K.; Sahu, P.; Kumar, P. Bacterial diversity in rhizosphere of Paspalum scrobiculatum L. (kodo millet) is revealed with shotgun metagenome sequencing and data analysis. Data Brief. 2018, 20, 1653–1657. [Google Scholar] [CrossRef]
- Chica, E.; Buela, L.; Valdez, A. Metagenomic survey of the bacterial communities in the rhizosphere of three Andean tuber crops. Symbiosis 2019, 1–10. [Google Scholar] [CrossRef]
- Bulgarelli, D. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, Y.K. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nat. Commun. 2017, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Prabha, R.; Gupta, V.K.; Verma, M.K. Metatranscriptome analysis deciphers multifunctional genes and enzymes linked with the degradation of aromatic compounds and pesticides in the wheat rhizosphere. Front. Microbiol. 2018, 9, 1331. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prabha, R.; Singh, D.P.; Gupta, S.; Gupta, V.K.; El-Enshasy, H.A.; Verma, M.K. Rhizosphere Metagenomics of Paspalum scrobiculatum L. (Kodo Millet) Reveals Rhizobiome Multifunctionalities. Microorganisms 2019, 7, 608. https://doi.org/10.3390/microorganisms7120608
Prabha R, Singh DP, Gupta S, Gupta VK, El-Enshasy HA, Verma MK. Rhizosphere Metagenomics of Paspalum scrobiculatum L. (Kodo Millet) Reveals Rhizobiome Multifunctionalities. Microorganisms. 2019; 7(12):608. https://doi.org/10.3390/microorganisms7120608
Chicago/Turabian StylePrabha, Ratna, Dhananjaya P. Singh, Shailendra Gupta, Vijai Kumar Gupta, Hesham A. El-Enshasy, and Mukesh K. Verma. 2019. "Rhizosphere Metagenomics of Paspalum scrobiculatum L. (Kodo Millet) Reveals Rhizobiome Multifunctionalities" Microorganisms 7, no. 12: 608. https://doi.org/10.3390/microorganisms7120608
APA StylePrabha, R., Singh, D. P., Gupta, S., Gupta, V. K., El-Enshasy, H. A., & Verma, M. K. (2019). Rhizosphere Metagenomics of Paspalum scrobiculatum L. (Kodo Millet) Reveals Rhizobiome Multifunctionalities. Microorganisms, 7(12), 608. https://doi.org/10.3390/microorganisms7120608