Site-Specific Microbial Decomposer Communities Do Not Imply Faster Decomposition: Results from a Litter Transplantation Experiment

 , ,
, ,  , and
, and 
Abstract
:
1. Introduction
2. Material and Methods
3. Results
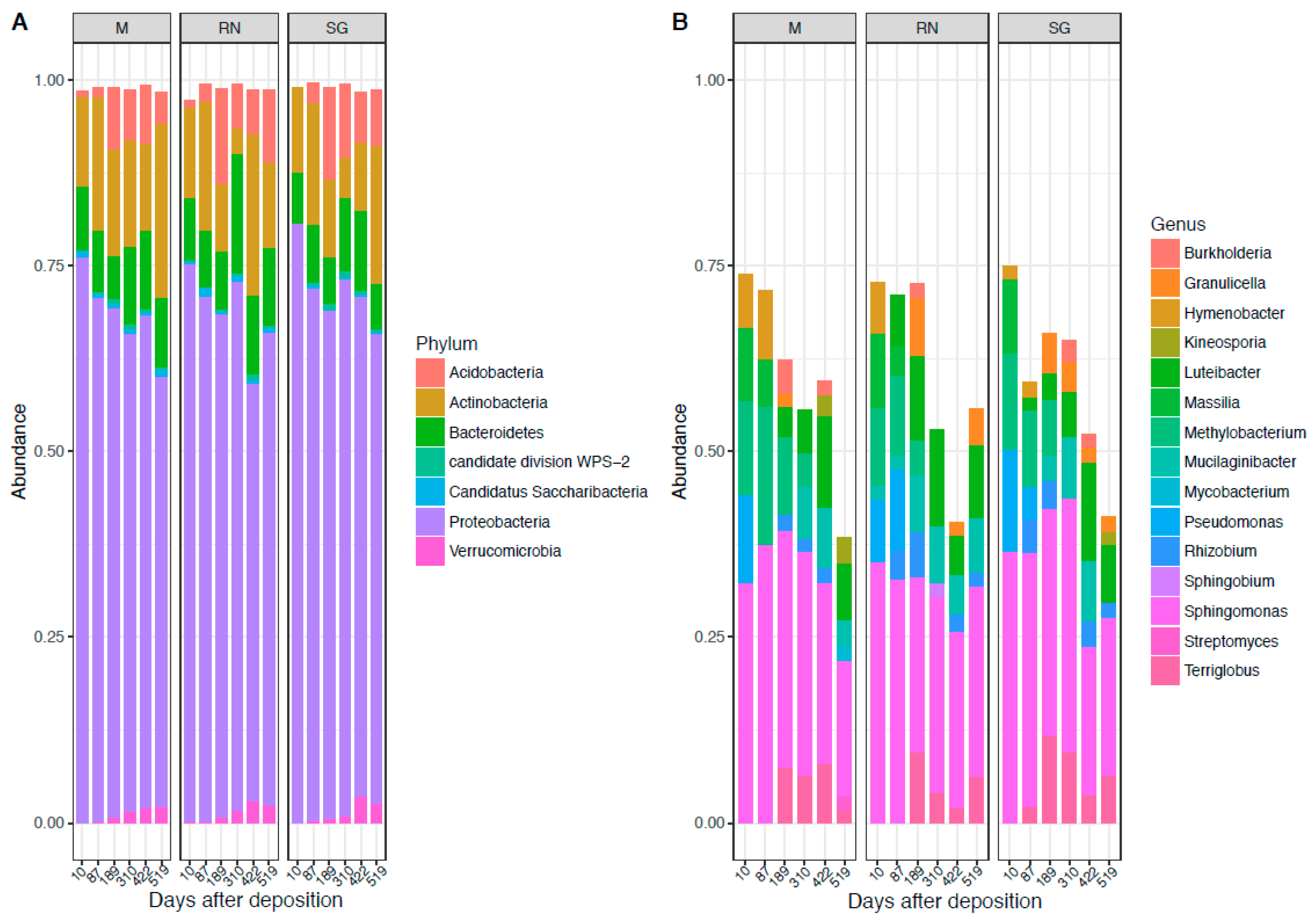
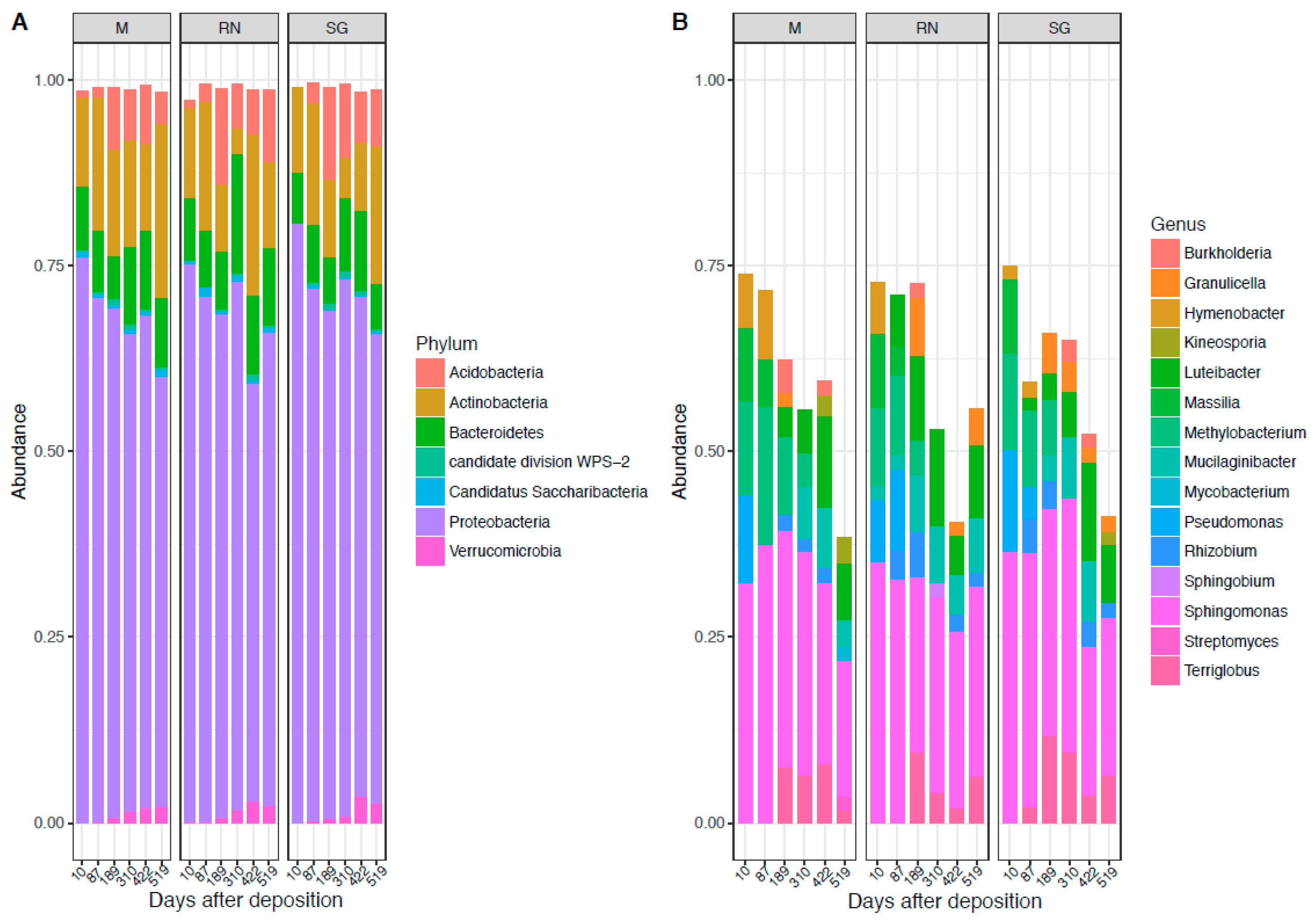
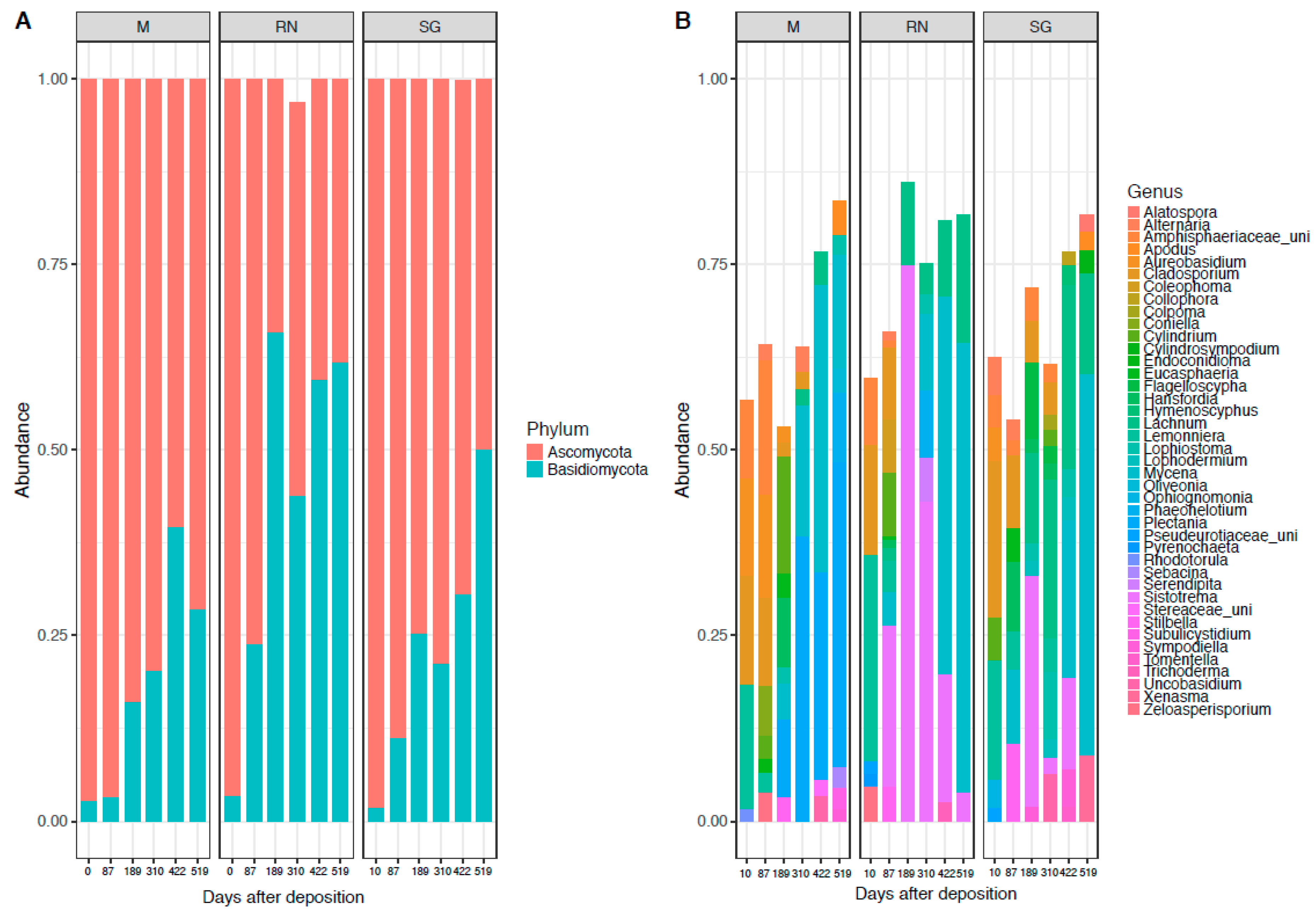
3.1. Bacterial and Fungal Community Composition
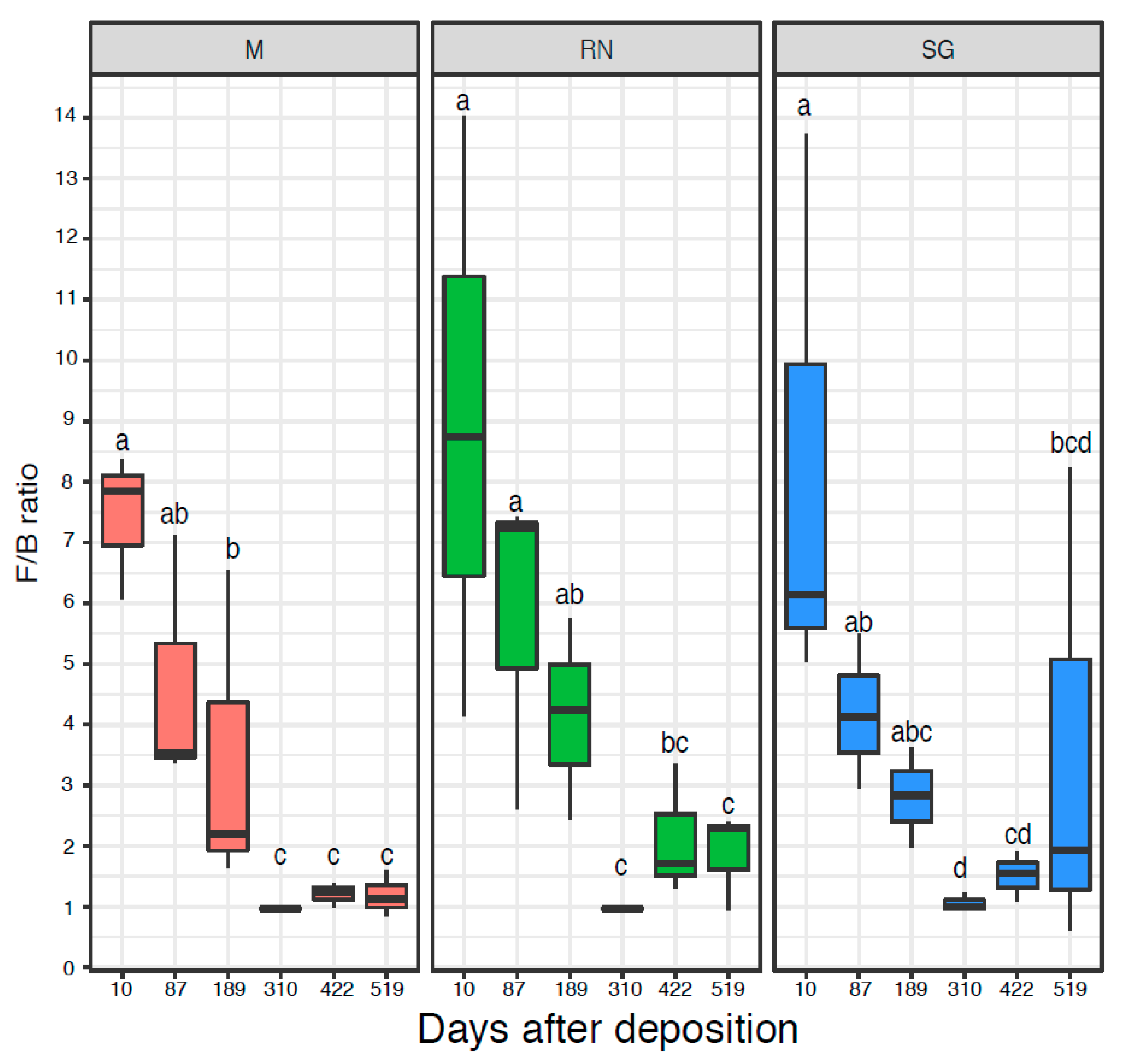
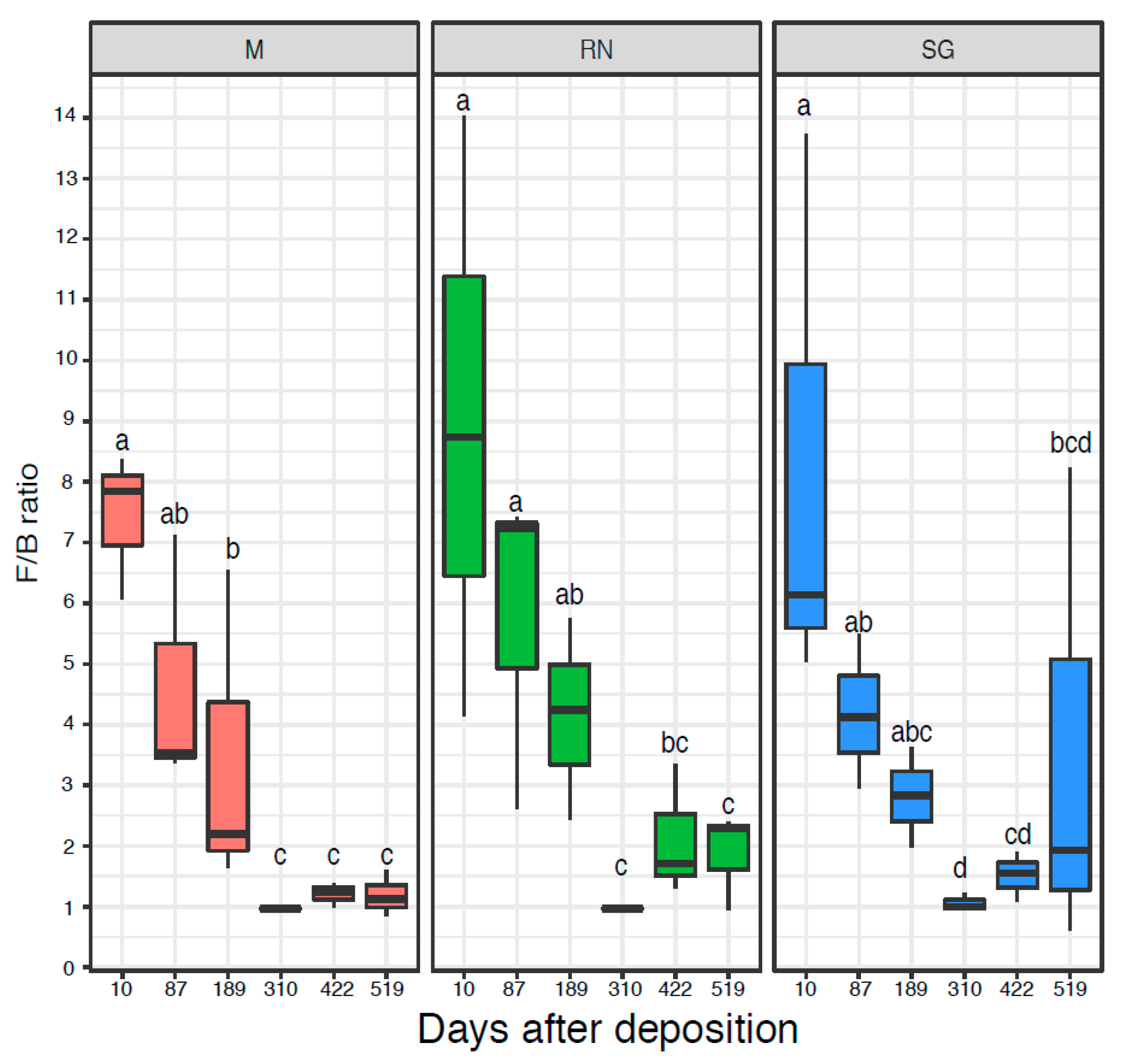
3.2. Bacterial and Fungal Quantification
3.3. Mass Loss and Litter Chemical Composition Dynamics
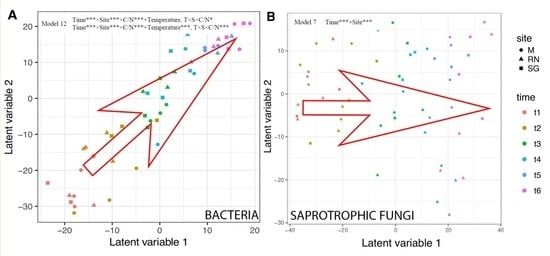
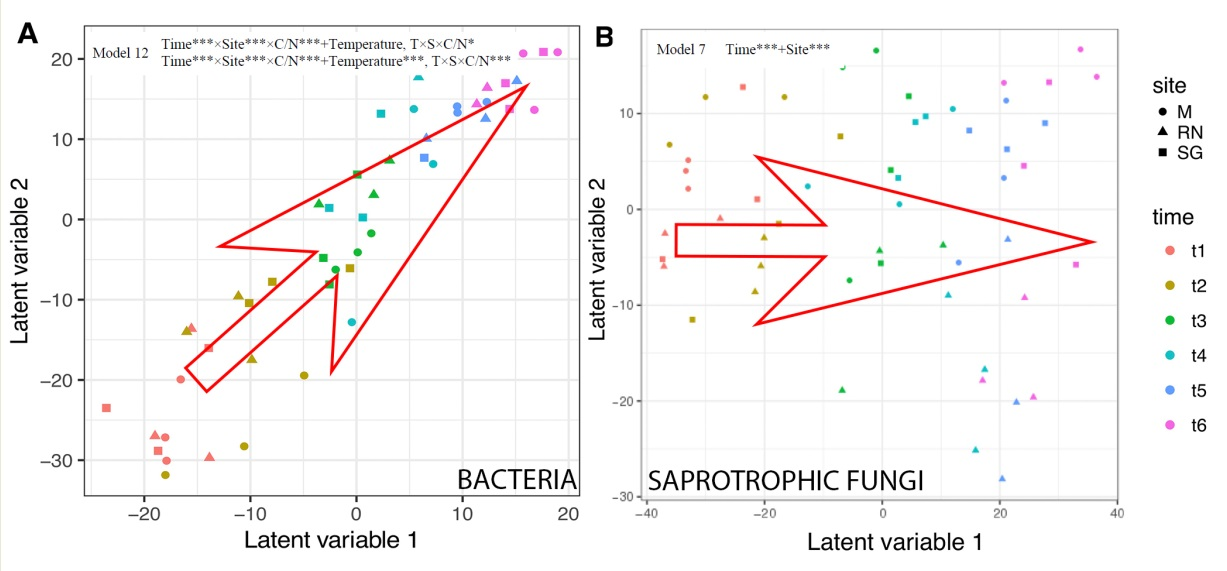
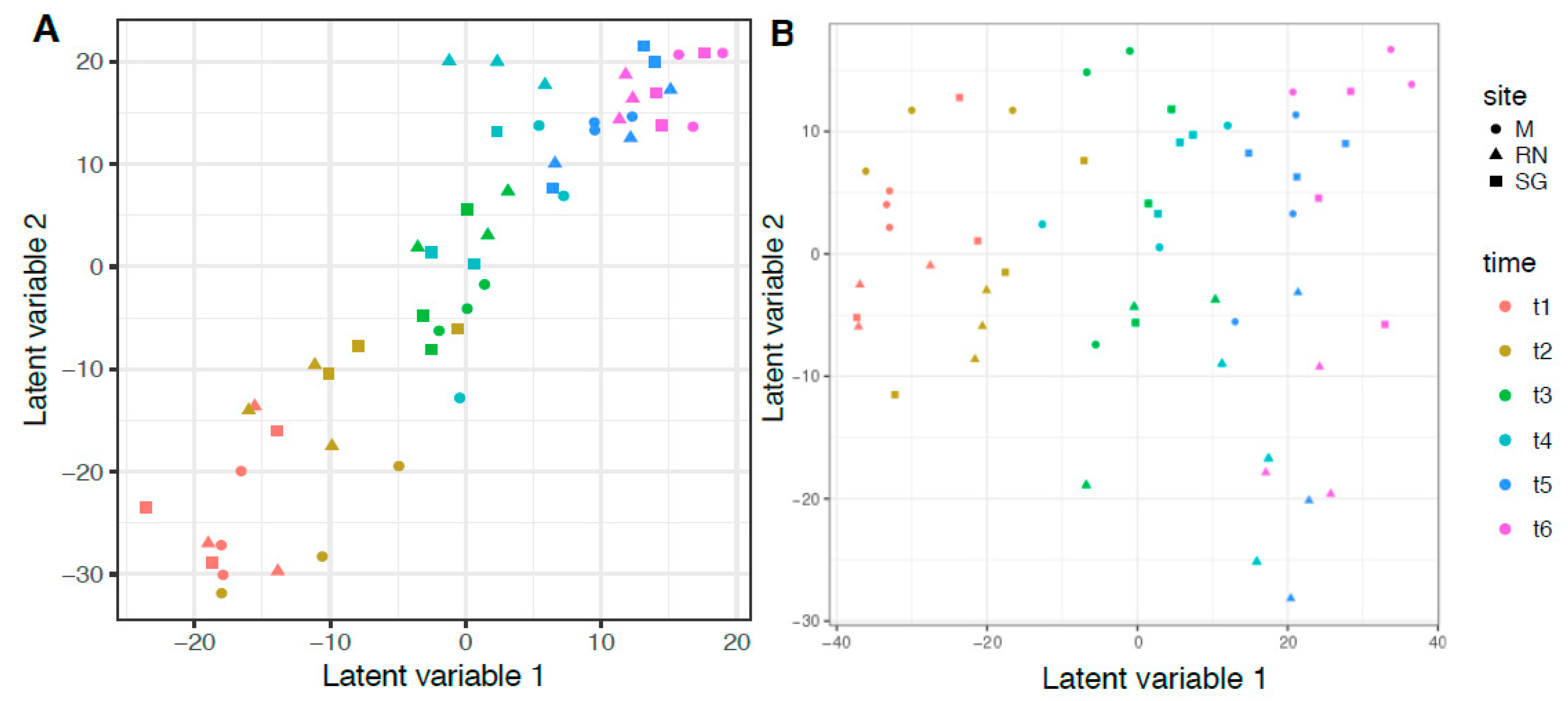
3.4. Bacterial and Fungal Community Structure
4. Discussion
4.1. Factors Explaining Microbial Composition
4.2. Microbial Community and Mass Loss Relationship
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindquist, E.J.; D’Annunzio, R.; Gerrand, A.; MacDicken, K.; Achard, F.; Beuchle, R.; Brink, A.; Eva, H.D.; Mayaux, P.; San-Miguel-Ayanz, J.; et al. Global Forest Land-Use Change 1990–2005; FAO: Rome, Italy, 2012. [Google Scholar]
- Didion, M.; Frey, B.; Rogiers, N.; Thürig, E. Validating tree litter decomposition in the Yasso07 carbon model. Ecol. Model. 2014, 291, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Osono, T.; Takeda, H. Potassium, calcium, and magnesium dynamics during litter decomposition in a cool temperate forest. J. For. Res. 2004, 9, 23–31. [Google Scholar] [CrossRef]
- Cotrufo, M.F.; De Angelis, P.; Polle, A. Leaf litter production and decomposition in a poplar short-rotation coppice exposed to free air CO2 enrichment (POPFACE). Glob. Chang. Biol. 2005, 11, 971–982. [Google Scholar] [CrossRef]
- Manzoni, S.; Trofymow, J.A.; Jackson, R.B.; Porporato, A. Stoichiometric controls on carbon, nitrogen, and phosphorus dynamics in decomposing litter. Ecol. Monogr. 2010, 80, 89–106. [Google Scholar] [CrossRef]
- Voříšková, J.; Brabcová, V.; Cajthaml, T.; Baldrian, P. Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol. 2014, 201, 269–278. [Google Scholar] [CrossRef] [PubMed]
- De Boer, W.; Folman, L.B.; Summerbell, R.C.; Boddy, L. Living in a fungal world: Impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 2005, 29, 795–811. [Google Scholar] [CrossRef] [PubMed]
- López-Mondéjar, R.; Zühlke, D.; Becher, D.; Riedel, K.; Baldrian, P. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 2016, 6, 25279. [Google Scholar] [CrossRef]
- Štursová, M.; Žifčáková, L.; Leigh, M.B.; Burgess, R.; Baldrian, P. Cellulose utilization in forest litter and soil: Identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 2012, 80, 735–746. [Google Scholar] [CrossRef]
- Probst, M.; Gómez-Brandón, M.; Bardelli, T.; Egli, M.; Insam, H.; Ascher-Jenull, J. Bacterial communities of decaying Norway spruce follow distinct slope exposure and time-dependent trajectories. Environ. Microbiol. 2018, 20, 3657–3670. [Google Scholar] [CrossRef]
- Ayres, E.; Steltzer, H.; Simmons, B.L.; Simpson, R.T.; Steinweg, J.M.; Wallenstein, M.D.; Mellor, N.; Parton, W.J.; Moore, J.C.; Wall, D.H. Home-field advantage accelerates leaf litter decomposition in forests. Soil Boil. Biochem. 2009, 41, 606–610. [Google Scholar] [CrossRef]
- Prescott, C.E.; Grayston, S.J. Tree species influence on microbial communities in litter and soil: Current knowledge and research needs. For. Ecol. Manag. 2013, 309, 19–27. [Google Scholar] [CrossRef]
- Veen, G.F.C.; Freschet, G.T.; Ordonez, A.; Wardle, D.A. Litter quality and environmental controls of home-field advantage effects on litter decomposition. Oikos 2015, 124, 187–195. [Google Scholar] [CrossRef]
- Aponte, C.; Garcia, L.V.; Marañón, T. Tree species effect on litter decomposition and nutrient release in Mediterranean oak forests changes over time. Ecosystems 2012, 15, 1204–1218. [Google Scholar] [CrossRef]
- Makkonen, M.; Berg, M.P.; Handa, I.T.; Hättenschwiler, S.; van Ruijven, J.; van Bodegom, P.M.; Aerts, R. Highly consistent effects of plant litter identity and functional traits on decomposition across a latitudinal gradient. Ecol. Lett. 2012, 15, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Bani, A.; Borruso, L.; Fornasier, F.; Pioli, S.; Wellstein, C.; Brusetti, L. Microbial decomposer dynamics: Diversity and functionality investigated through a transplantation experiment in boreal forests. Microb. Ecol. 2018, 76, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, L.; Austin, A. Tree species identity alters forest litter decomposition through long-term plant and soil interactions in Patagonia, Argentina. J. Ecol. 2008, 96, 727–736. [Google Scholar] [CrossRef]
- Palozzi, J.E.; Lindo, Z. Are leaf litter and microbes team players? Interpreting home-field advantage decomposition dynamics. Soil Biol. Biochem. 2018, 124, 189–198. [Google Scholar] [CrossRef]
- Aneja, M.K.; Sharma, S.; Fleischmann, F.; Stich, S.; Heller, W.; Bahnweg, G.; Munch, J.C.; Schloter, M. Microbial Colonization of Beech and Spruce Litter—Influence of Decomposition Site and Plant Litter Species on the Diversity of Microbial Community. Microb. Ecol. 2006, 52, 127–135. [Google Scholar] [CrossRef]
- Veen, G.F.C.; Sundqvist, M.K.; Wardle, D.A. Environmental factors and traits that drive plant litter decomposition do not determine home-field advantage effects. Funct. Ecol. 2015, 29, 981–991. [Google Scholar] [CrossRef]
- López-Mondéjar, R.; Voříšková, J.; Větrovský, T.; Baldrian, P. The bacterial community inhabiting temperate deciduous forests is vertically stratified and undergoes seasonal dynamics. Soil Biol. Biochem. 2015, 87, 43–50. [Google Scholar] [CrossRef]
- Das, S.; Dash, H.R. Microbial Diversity in the Genomic Era; Academic Press: Cambridge, MA, USA, 2018; pp. 3–9. [Google Scholar]
- Cornelissen, J.H.C.; Lavorel, S.; Garnier, E.; Díaz, S.; Buchmann, N.; Gurvich, D.E.; Reich, P.B.; ter Steege, H.; Morgan, H.D.; van der Heijden, M.G.A.; et al. A handbook of protocols for standardised and easy measurement of plant functional traits worldwide. Aus. J. Bot. 2003, 51, 335–380. [Google Scholar] [CrossRef] [Green Version]
- Ferris, M.J.; Muyzer, G.; Ward, A.D.M. Denaturing Gradient Gel Electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 1996, 62, 340–346. [Google Scholar] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Appl. Lab. Man. 1990, 18, 315–322. [Google Scholar]
- Bardelli, T.; Gómez-Brandón, M.; Ascher-Jenull, J.; Fornasier, F.; Arfaioli, P.; Francioli, D.; Egli, M.; Sartori, G.; Insam, H.; Pietramellara, G. Effects of slope exposure on soil physico-chemical and microbiological properties along an altitudinal climosequence in the Italian Alps. Sci. Total. Environ. 2017, 575, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- De Beeck, M.O.; Lievens, B.; Busschaert, P.; Declerck, S.; Vangronsveld, J.; Colpaert, J.V. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE 2014, 9, e97629. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumbrell, A.J.; Ferguson, R.M.W.; Clark, D.R. Hydrocarbon and Lipid Microbiology Protocols; Springer: Berlin/Heidelberg, Germany, 2017; pp. 155–206. [Google Scholar]
- Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for Fastq Files, Version 1.33. Software. 2011. Available online: https://github.com/najoshi/sickle (accessed on 26 January 2019).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Boil. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013, 30, 614. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. Peer J. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Callaghan, T.M.; Douglas, B.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Snipes, M.; Taylor, D.C. Model selection and Akaike Information Criteria: An example from wine ratings and prices. Wine Econ. Policy 2014, 3, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression Second; Sage: Thousand oaks, CA, USA, 2011. [Google Scholar]
- De Mendiburu, F. Package ‘Agricolae.’ R Package Version 1.2-8. Available online: http://CRAN.R-project.org/package=agricolae (accessed on 4 July 2019).
- McMurdie, P.J.; Holmes, S. Phyloseq: A R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Naumann, U.; Wright, S.T.; Warton, D.I. Mvabund–an R package for model-based analysis of multivariate abundance data. Methods Ecol. Evol. 2012, 3, 471–474. [Google Scholar] [CrossRef]
- Hui, F.K.C. Boral–Bayesian ordination and regression analysis of multivariate abundance data in R. Methods Ecol. Evol. 2016, 7, 744–750. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. Corrplot: Visualization of a Correlation Matrix. R Package Version 0.77. 2016. Available online: https://cran.microsoft.com/snapshot/2016-08-01/web/packages/corrplot/index.html (accessed on 21 April 2016).
- Hmisc: Harrell Miscellaneous, version 3.12-2; Computer Software; 2013. Available online: http://cran.R-project.Org/web/packages/Hmisc (accessed on 26 January 2019).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Bálint, M.; Bartha, L.; O’Hara, R.B.; Olson, M.S.; Otte, J.; Pfenninger, M.; Robertson, A.L.; Tiffin, P.; Schmitt, L. Relocation, high-latitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol. Ecol. 2015, 24, 235–248. [Google Scholar] [CrossRef]
- Purahong, W.; Kapturska, D.; Pecyna, M.J.; Jariyavidyanont, K.; Kaunzner, J.; Juncheed, K.; Uengwetwanit, T.; Rudloff, R.; Schulz, E.; Hofrichter, M.; et al. Effects of Forest Management Practices in Temperate Beech Forests on Bacterial and Fungal Communities Involved in Leaf Litter Degradation. Microb. Ecol. 2015, 69, 905–913. [Google Scholar] [CrossRef]
- Purahong, W.; Wubet, T.; Lentendu, G.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Hofrichter, M.; Krüger, D.; Buscot, F. Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 2016, 25, 4059–4074. [Google Scholar] [CrossRef]
- Bani, A.; Pioli, S.; Ventura, M.; Panzacchi, P.; Borruso, L.; Tognetti, R.; Tonon, G.; Brusetti, L. The role of microbial community in the decomposition of leaf litter and deadwood. Appl. Soil Ecol. 2018, 126, 75–84. [Google Scholar] [CrossRef]
- Pioli, S.; Antonucci, S.; Giovannelli, A.; Traversi, M.L.; Borruso, L.; Bani, A.; Brusetti, L.; Tognetti, R. Community fingerprinting reveals increasing wood-inhabiting fungal diversity in unmanaged Mediterranean forests. For. Ecol. Manag. 2018, 408, 202–210. [Google Scholar] [CrossRef]
- Van der Heijden, M.G.A.; de Bruin, S.; Luckerhoff, L.; van Logtestijn, R.S.P.; Schlaeppi, K. A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 2016, 10, 389–399. [Google Scholar] [CrossRef]
- McGuire, K.L.; Treseder, K.K. Microbial communities and their relevance for ecosystem models: Decomposition as a case study. Soil Biol. Biochem. 2010, 42, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Ovaskainen, O.; Hottola, J.; Siitonen, J. Modeling species co-occurrence by multivariate logistic regression generates new hypotheses on fungal interactions. Ecology 2010, 91, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Dickie, I.A.; Fukami, T.; Wilkie, J.P.; Allen, R.B.; Buchanan, P.K. Do assembly history effects attenuate from species to ecosystem properties? A field test with wood-inhabiting fungi. Ecol. Lett. 2012, 15, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.M.; Bruns, T.D.; Taylor, J.W.; Smith, D.P.; Branco, S.; Glassman, S.I.; Erlandson, S.; Vilgalys, R.; Liao, H.L.; Smith, M.E.; et al. Endemism and functional convergence across the North American soil mycobiome. Proc. Natl. Acad. Sci. USA 2014, 111, 6341–6346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dix, N.J.; Webster, J. Fungal Ecology; Chapman & Hall: London, UK, 1995. [Google Scholar]
- Miyamoto, T. Lignin-degrading ability of litter-decomposing basidiomycetes from Picea forests of Hokkaido. Mycoscience 2000, 41, 105–110. [Google Scholar] [CrossRef]
- Tamames, J.; Abellán, J.J.; Pignatelli, M.; Camacho, A.; Moya, A. Environmental distribution of prokaryotic taxa. BMC Microbiol. 2010, 10, 85. [Google Scholar] [CrossRef]
- Polo, A.; Cappitelli, F.; Brusetti, L.; Principi, P.; Villa, F.; Giacomucci, L.; Ranalli, G.; Sorlini, C. Feasibility of Removing Surface Deposits on Stone Using Biological and Chemical Remediation Methods. Microb. Ecol. 2010, 60, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nogales, J.; García, J.L.; Díaz, E. Degradation of Aromatic Compounds in Pseudomonas: A Systems Biology View. In Aerobic Utilization of Hydrocarbons, Oils and Lipids. Handbook of Hydrocarbon and Lipid Microbiology; Rojo, F., Ed.; Springer: Cham, Switzerland, 2017; pp. 1–49. [Google Scholar]
- Ayres, E.; Dromph, K.M.; Bardgett, R.D. Do plant species encourage soil biota that specialise in the rapid decomposition of their litter? Soil Biol. Biochem. 2006, 38, 183–186. [Google Scholar] [CrossRef]
- Asplund, J.; Kauserud, H.; Bokhorst, S.; Lie, M.H.; Ohlson, M.; Nybakken, L. Fungal communities influence decomposition rates of plant litter from two dominant tree species. Fungal Ecol. 2018, 32, 1–8. [Google Scholar] [CrossRef]
- Purahong, W.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Däumlich, V.; Mital, S.; Buscot, F.; Hofrichter, M.; Gutknecht, J.L.M.; Krüger, D. Uncoupling of microbial community structure and function in decomposing litter across beech forest ecosystems in Central Europe. Sci. Rep. 2014, 4, 7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagler, M.; Ascher, J.; Gómez-Brandón, M.; Insam, H. Soil microbial communities along the route of a venturous cycling trip. Appl. Soil Ecol. 2016, 99, 13–18. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Characteristic | Monticolo | San Genesio Atesino | Renon |
|---|---|---|---|
| Elevation | 530 m a.s.l. | 1000 m a.s.l. | 1530 m a.s.l. |
| GPS coordinates | 46°25′35″ N; 11°17′55″ E | 46°32′35.4″ N; 11°18′36.4″ E | 46°35′07.1″ N; 11°25′44.4″ E |
| Lithology | Quartz porphyritic | Quartz porphyritic | Quartz porphyritic |
| Soil type | Acid brown soil | Podzol | Podzol |
| Soil texture | Sandy loam | Sandy loam | Sandy loam |
| pH soil | 5.50 | 6.13 | 4.83 |
| Mean annual precipitation | 800 mm | 735 mm | 970 mm |
| Mean annual temperature | 11.8 °C (max. 21.3 °C, min. 3.6 °C) | 10.5 °C (max. 17.0 °C, min. 3.0 °C) | 6.8 °C (max. 14.3 °C, min. −0.7 °C) |
| Dominant vegetation | Oak (Quercus petraea) | European beech (Fagus sylvatica) | Rhododendron (alpine rose) (Rhododendron ferrugineum) |
| Trophic Mode | Number of Genera | Relative Frequencies |
|---|---|---|
| Pathotroph (Pat) | 66 | 7.810245 |
| Saprotroph (Sa) | 104 | 53.52034 |
| Symbiotroph (Sy) | 26 | 9.91289 |
| Pathotroph-Saprotroph (Pat-Sa) | 10 | 0.8716193 |
| Pathotroph-Symbiotroph (Pat-Sy) | 8 | 1.627169 |
| Saprotroph-Symbiotroph (Sa-Sy) | 8 | 15.23502 |
| Pathogen-Saprotroph-Symbiotroph (Pa-Sa-Sy) | 1 | 0.03621281 |
| Pathotroph-Saprotroph-Symbiotroph (Pat-Sa-Sy) | 14 | 4.793488 |
| Unassigned (U) | 35 | 1.621936 |
| Total | 320 | 100 |
| Model. | Variables | AIC Score Bacteria | AIC Score Saprotrophic |
|---|---|---|---|
| Null | 1 | 46,568 | 28,094 |
| Model 1 | Time *** | 43,920 | 27,094 |
| Model 2 | Site *** | 46,530 | 27,963 |
| Model 3 | Total N *** | 44,252 | 27,272 |
| Model 4 | Total C *** | 46,031 | 33,242 |
| Model 5 | C/N ratio *** | 44,126 | 27,272 |
| Model 6 | Temperature *** | 45,989 | 33,065 |
| Model 7 | Time*** + Site *** | 43,744 | 26,908 |
| Model 8 | Time *** × Site ** (Time × Site) *** | 43,753 | 27,019 |
| Model 9 | Time *** × Site ** × Temperature *** | 46,927 | 38,792 |
| Model 10 | Time *** × Site *** × C/N *** | 42,351 | 35,163 |
| Model 11 | Time *** + Site *** + C/N *** + Temperature Time *** + Site *** + C/N *** + Temperature *** | 43,160 | 30,818 |
| Model 12 | Time *** × Site *** × C/N *** + Temperature, T × S × C/N * Time *** × Site *** × C/N *** + Temperature ***, T × S × C/N *** | 42,558 | 35,537 |
| Model 13 | Time *** + Site *** + TotalC ** + TotalN ** + C/N *** + Temperature *** Time *** + Site *** + TotalC *** + TotalN *** + C/N ** + Temperature *** | 43,268 | 150,575 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bani, A.; Borruso, L.; Matthews Nicholass, K.J.; Bardelli, T.; Polo, A.; Pioli, S.; Gómez-Brandón, M.; Insam, H.; Dumbrell, A.J.; Brusetti, L. Site-Specific Microbial Decomposer Communities Do Not Imply Faster Decomposition: Results from a Litter Transplantation Experiment. Microorganisms 2019, 7, 349. https://doi.org/10.3390/microorganisms7090349
Bani A, Borruso L, Matthews Nicholass KJ, Bardelli T, Polo A, Pioli S, Gómez-Brandón M, Insam H, Dumbrell AJ, Brusetti L. Site-Specific Microbial Decomposer Communities Do Not Imply Faster Decomposition: Results from a Litter Transplantation Experiment. Microorganisms. 2019; 7(9):349. https://doi.org/10.3390/microorganisms7090349
Chicago/Turabian StyleBani, Alessia, Luigimaria Borruso, Kirsty J. Matthews Nicholass, Tommaso Bardelli, Andrea Polo, Silvia Pioli, María Gómez-Brandón, Heribert Insam, Alex J. Dumbrell, and Lorenzo Brusetti. 2019. "Site-Specific Microbial Decomposer Communities Do Not Imply Faster Decomposition: Results from a Litter Transplantation Experiment" Microorganisms 7, no. 9: 349. https://doi.org/10.3390/microorganisms7090349
APA StyleBani, A., Borruso, L., Matthews Nicholass, K. J., Bardelli, T., Polo, A., Pioli, S., Gómez-Brandón, M., Insam, H., Dumbrell, A. J., & Brusetti, L. (2019). Site-Specific Microbial Decomposer Communities Do Not Imply Faster Decomposition: Results from a Litter Transplantation Experiment. Microorganisms, 7(9), 349. https://doi.org/10.3390/microorganisms7090349