Temporal Variability of Virioplankton during a Gymnodinium catenatum Algal Bloom



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Environmental Parameters
2.2. Collection of Environmental DNA
2.3. Metagenomic Sequencing
2.4. Data Analysis
2.5. Data Availability
3. Results and Discussion
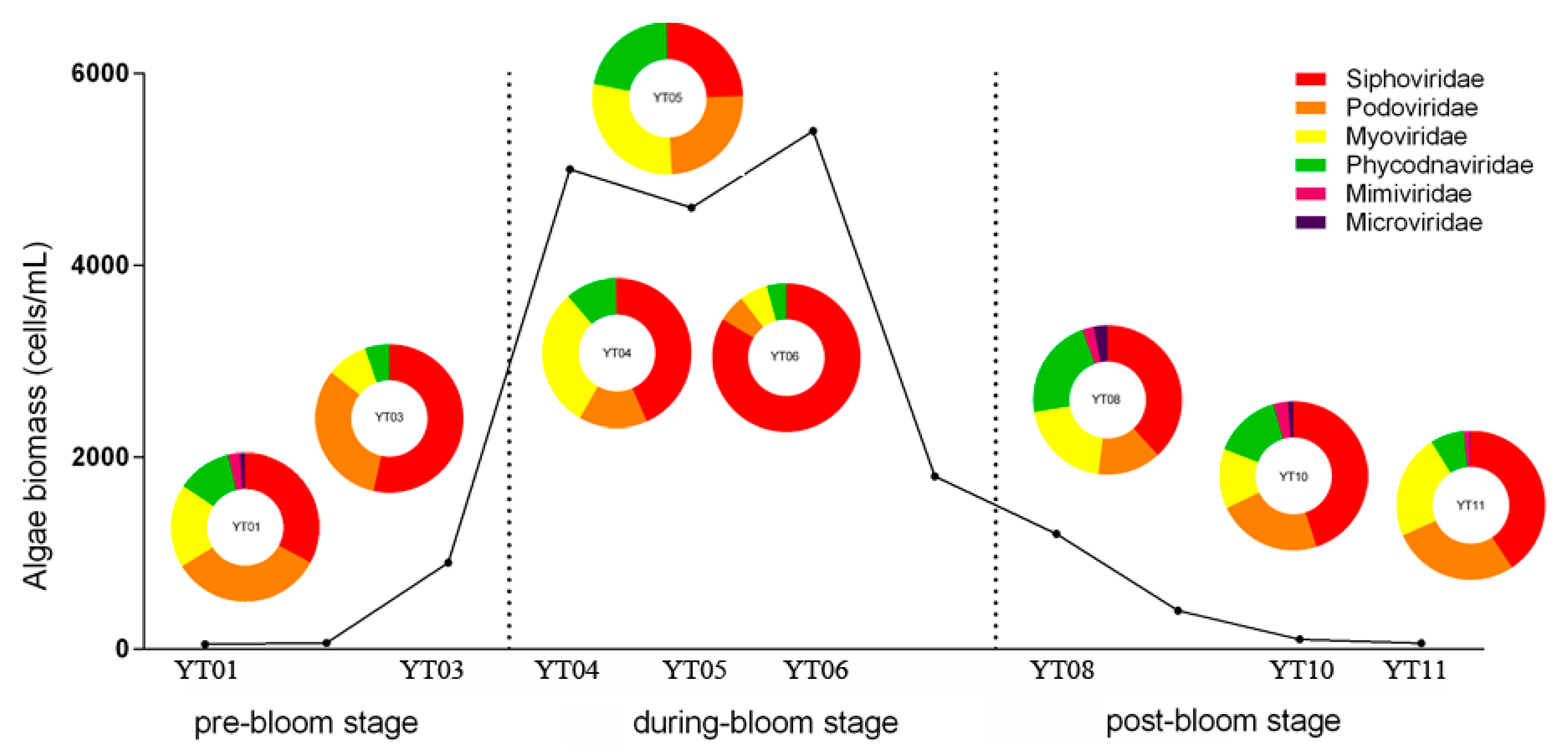
3.1. Algal Bloom Characteristics
3.2. Metagenomic Data
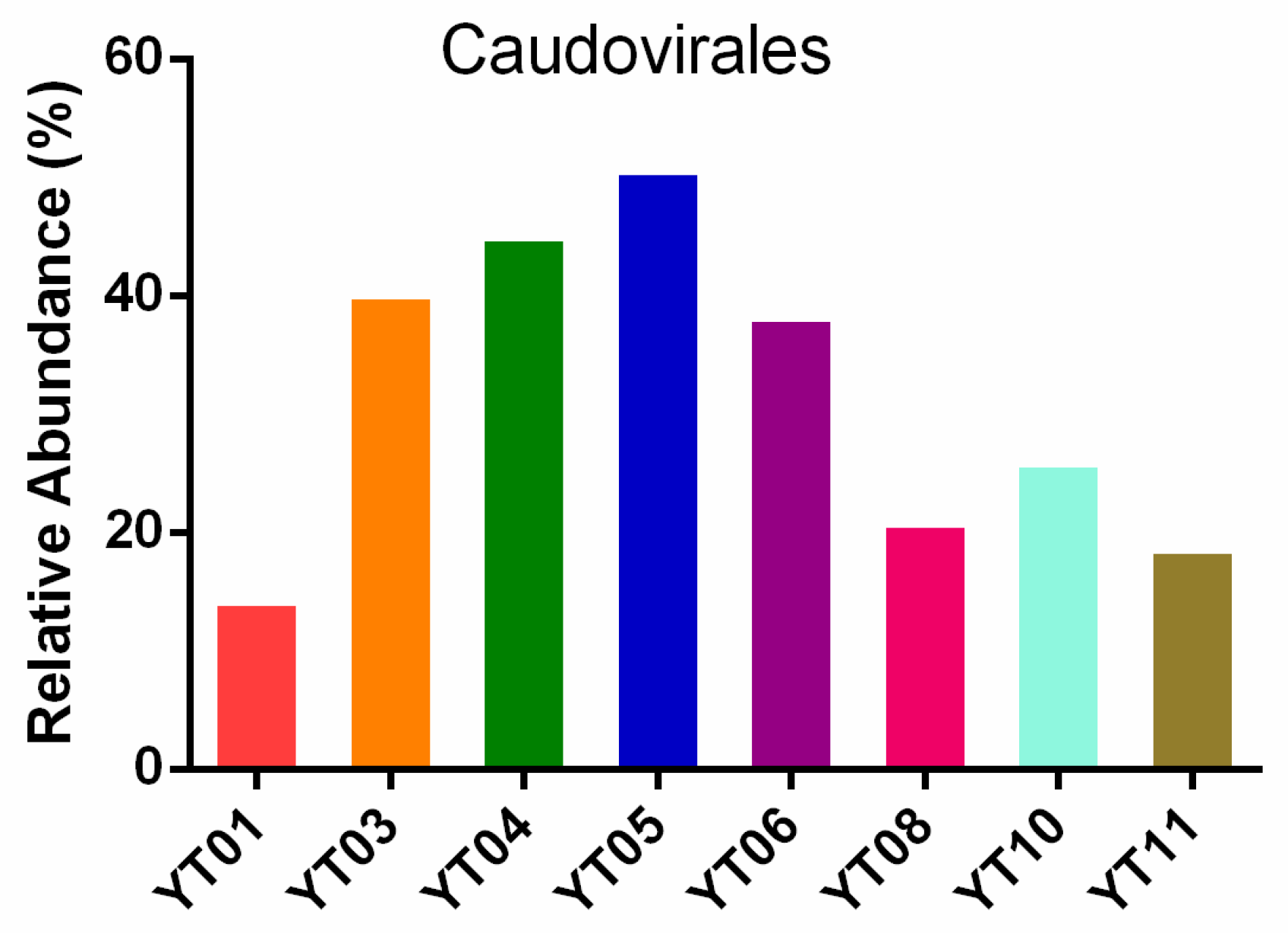
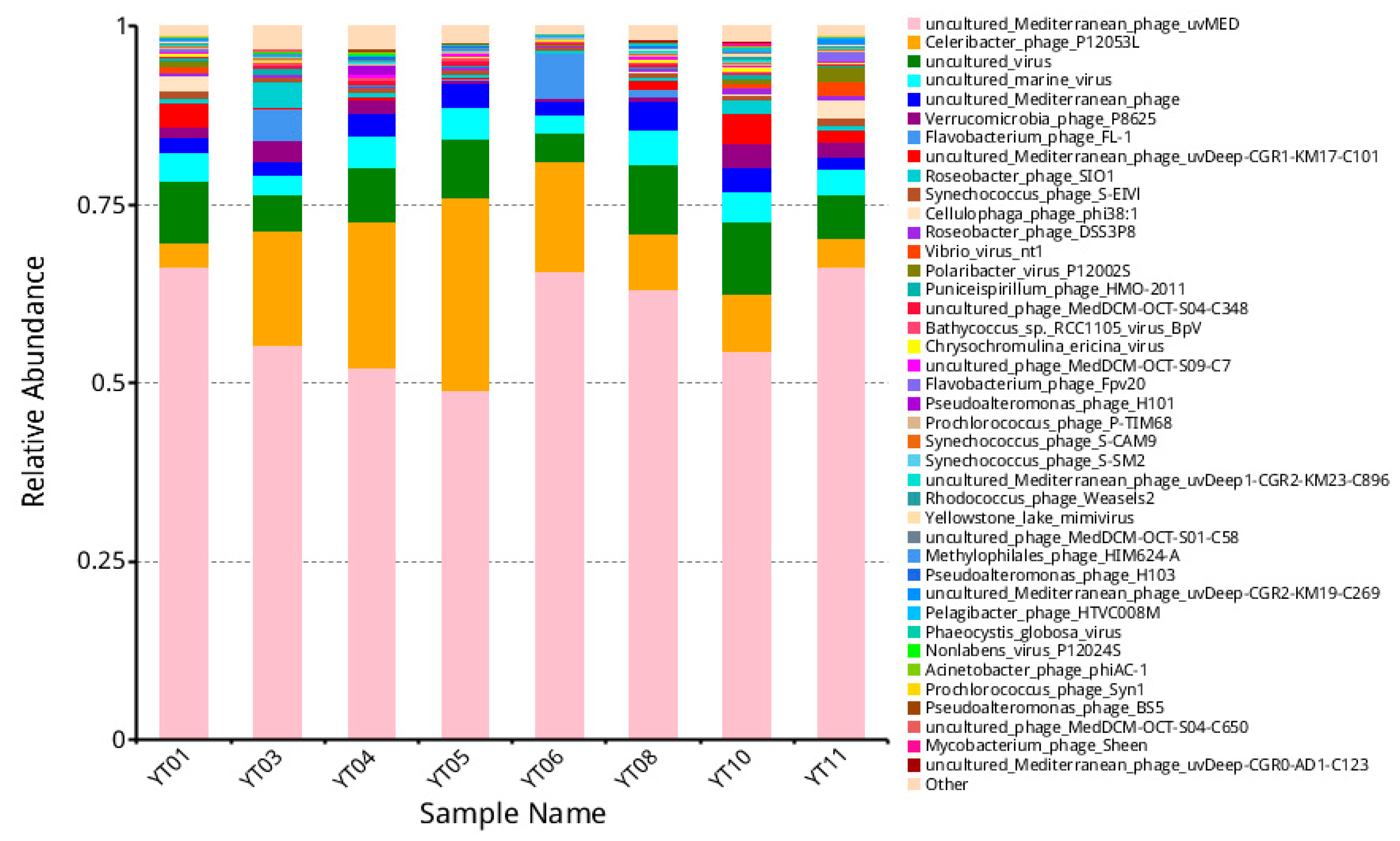
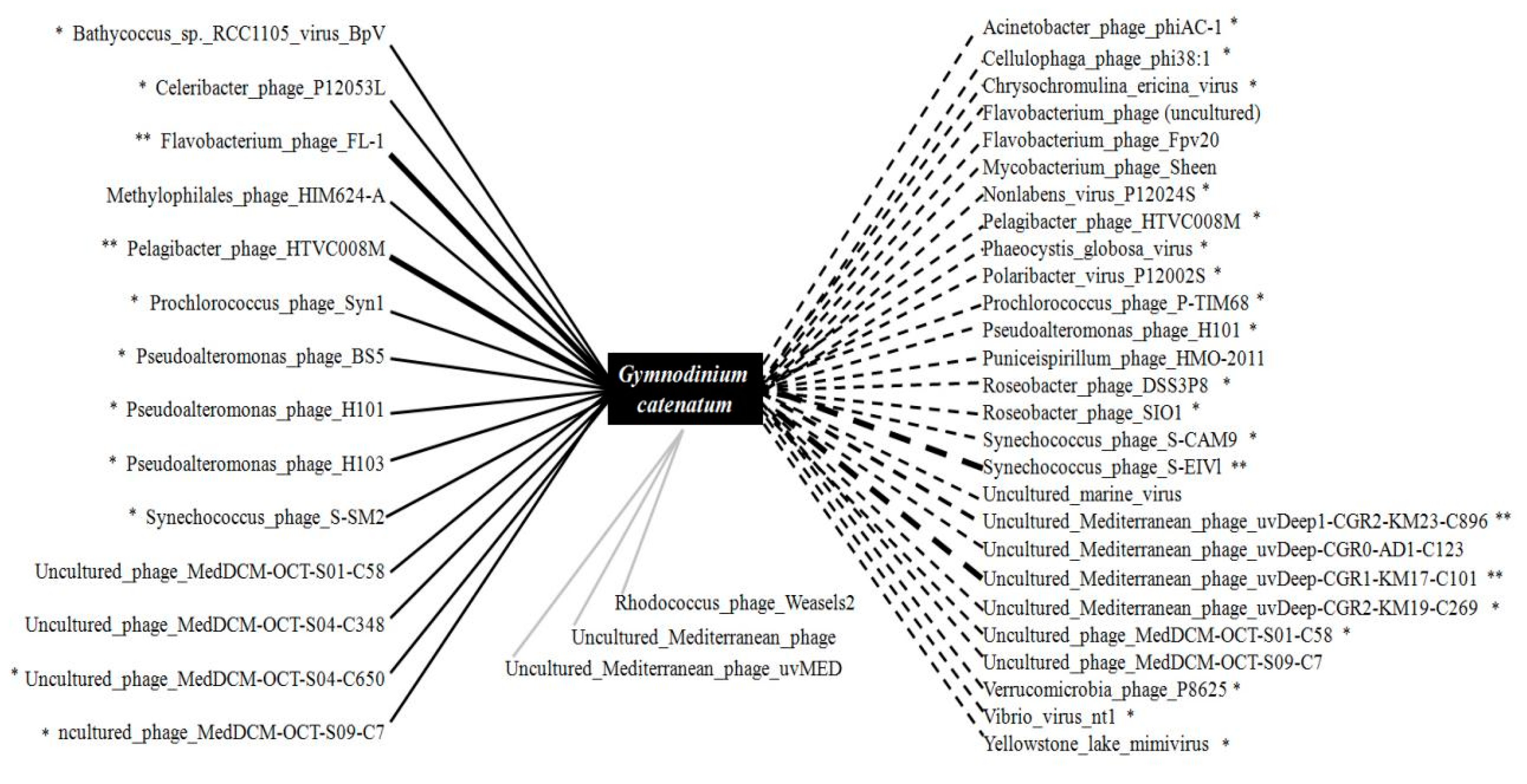
3.3. Taxonomic Composition
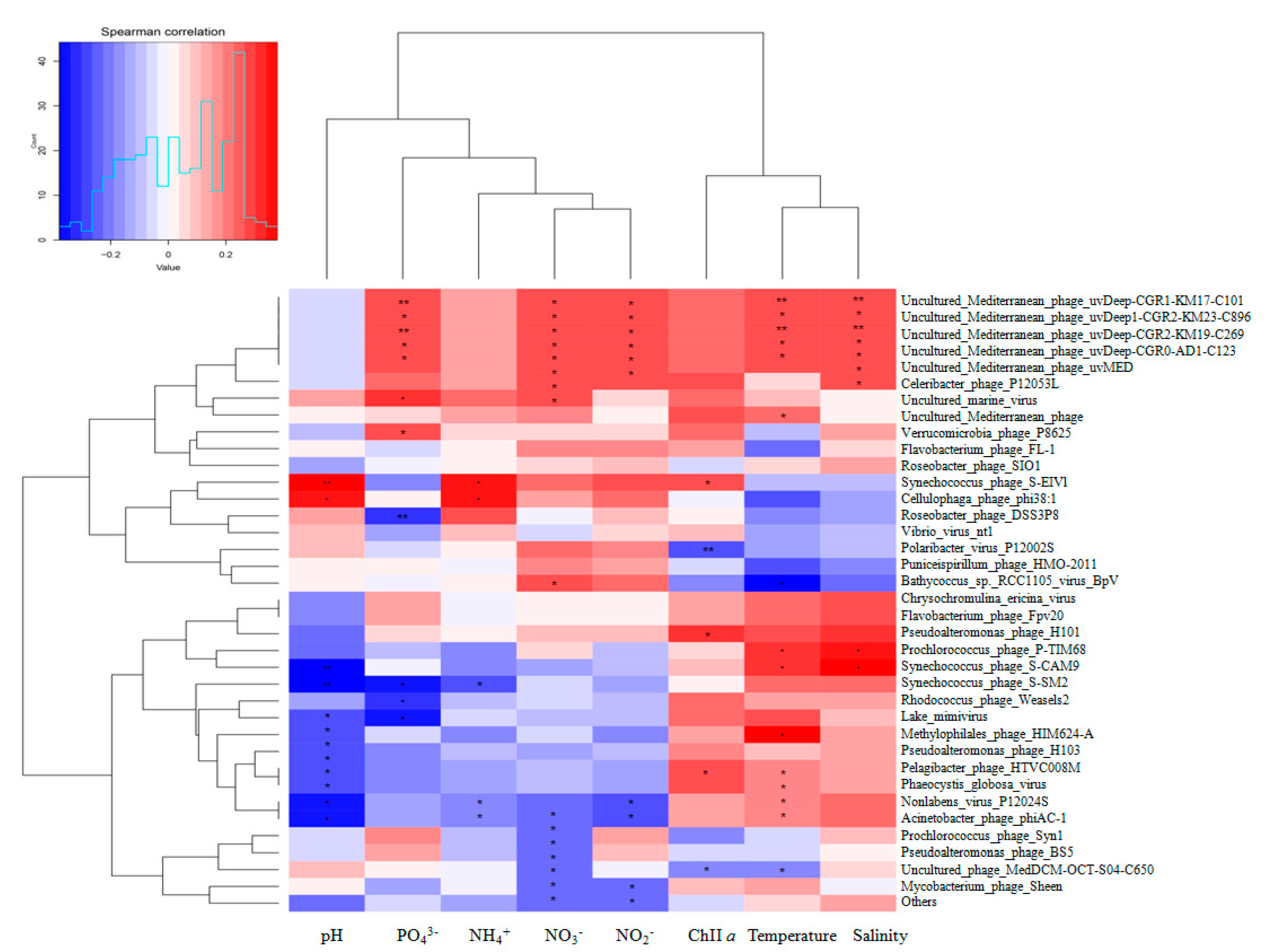
3.4. Associations between Viral Communities and Environmental Variables
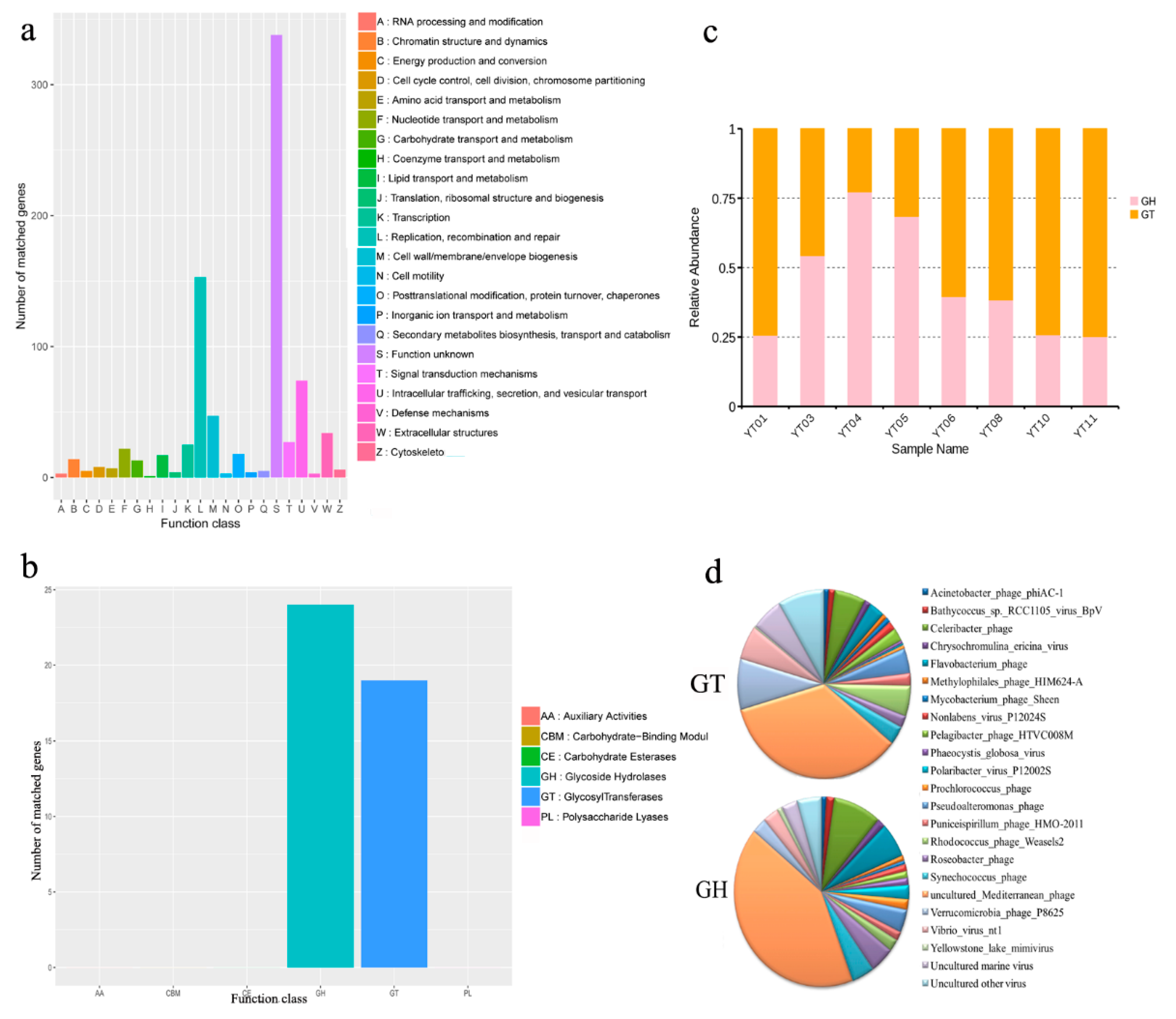
3.5. Functional Prediction of Algal Bloom Sample Viromes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Field, C.B.; Behrenfeld, M.J.; Randerson, J.T.; Falkowski, P. Primary production of the biosphere: Integrating terrestrial and oceanic components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Behrenfeld, M.J.; O’Malley, R.T.; Siegel, D.A.; McClain, C.R.; Sarmiento, J.L.; Feldman, G.C.; Milligan, A.J.; Falkowski, P.G.; Letelier, R.M.; Boss, E.S. Climate-driven trends in contemporary ocean productivity. Nature 2006, 444, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Shi, K.; Liu, J.J.; Deng, J.M.; Qin, B.Q.; Zhu, G.W.; Zhou, Y.Q. Meteorological and hydrological conditions driving the formation and disappearance of black blooms, an ecological disaster phenomena of eutrophication and algal blooms. Sci. Total. Environ. 2016, 569, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Cembella, A.D.; Hallegraeff, G.M. Progress in understanding harmful algal blooms: Paradigm shifts and new technologies for research, monitoring, and management. Ann. Rev. Mar. Sci. 2012, 4, 143–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdalet, E.; Fleming, L.E.; Gowen, R.; Davidson, K.; Hess, P.; Backer, L.C.; Moore, S.K.; Hoagland, P.; Enevoldsen, H. Marine harmful algal blooms, human health and well being: Challenges and opportunities in the 21st century. J. Mar. Biol. Assoc. UK 2016, 96, 61–91. [Google Scholar] [CrossRef] [Green Version]
- Heisler, J.; Glibert, P.M.; Burkholder, J.M.; Anderson, D.M.; Cochlan, W.; Dennison, W.C.; Dortch, Q.; Gobler, C.J.; Heil, C.A.; Humph-ries, E.; et al. Eutrophication and harmful algal blooms: A scientific consensus. Harmful Algae 2008, 8, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Murray, S.A.; Wiese, M.; Stüken, A.; Brett, S.; Kellmann, R.; Hallegraeff, G.; Neilan, B.A. SxtA-based quantitative molecular assay to identify saxitoxin-producing harmful algal blooms in marine waters. Appl. Environ. Microb. 2011, 77, 7050–7057. [Google Scholar] [CrossRef] [Green Version]
- Shunmugam, S.; Jokela, J.; Wahlsten, M.; Battchikova, N.; Rehman, A.U.; Vass, I.; Karonen, M.; Sinkkonen, J.; Permi, P.; Sivonen, K. Secondary metabolite from Nostoc XPORK14A inhibits photosynthesis and growth of synecho cyst is PCC6803. Plant Cell Environ. 2014, 37, 1371–1381. [Google Scholar] [CrossRef]
- Chen, Z.R.; Lei, X.B.; Zhang, B.Z.; Yang, L.X.; Zhang, J.H.; Zhang, J.Y.; Li, Y.; Zheng, W.; Tian, Y.; Liu, J.; et al. First Report of Pseudobodo sp, a new pathogen for a potential energy-producing algae: Chlorella vulgaris cultures. PLoS ONE 2014, 9, e89571. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhu, H.; Zhang, H.J.; Chen, Z.R.; Tian, Y.; Xu, H.; Zheng, T.L.; Zheng, W. Toxicity of algicidal extracts from Mangrovimonas yunxiaonensis strain LY01 on a HAB causing Alexandrium tamarense. J. Hazard Mater. 2014, 278, 372–381. [Google Scholar] [CrossRef]
- Cai, W.W.; Wang, H.; Tian, Y.; Chen, F.; Zheng, T.L. Influence of a bacteriophage on the population dynamics of toxic dinoflagellates by lysis of algicidal bacteria. Appl. Environ. Microb. 2011, 77, 7837–7840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucette, G.J. Interactions between bacteria and harmful algae: A review. Nat. Toxins 1995, 3, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, M.; Martin, J.L.; Rooney-Varga, J.N. Stimulation of Alexandrium fundyense growth by bacterial assemblages from the Bay of Fundy. J. Appl. Microbiol. 2002, 92, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segev, E.; Wyche, T.P.; Kim, K.H.; Petersen, J.; Ellebrandt, C.; Vlamakis, H.; Barteneva, N.; Paulson, J.N.; Chai, L.; Clardy, J.; et al. Dynamic metabolic exchange governs a marine algal-bacterial interaction. eLife 2016, 5, e17473. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Kanno, T.; Okamoto, R.; Itakura, S.; Yamaguchi, M.; Nishijima, T. Population structure of Alexandrium (Dinophyceae) cyst formation-promoting bacteria in Hiroshima Bay, Japan. Appl. Environ. Microbiol. 2003, 69, 6560–6568. [Google Scholar] [CrossRef] [Green Version]
- Demuez, M.; González-Fernández, C.; Ballesteros, M. Algicidal microorganisms and secreted algicides: New tools to induce microalgal cell disruption. Biotechnol. Adv. 2015, 33, 1615–1625. [Google Scholar] [CrossRef]
- Bloh, A.H.; Usup, G.; Ahmad, A. Loktanella spp. Gb03 as an algicidal bacterium, isolated from the culture of dinoflagellate Gambierdiscus belizeanus. Vet. World 2016, 9, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Sanders, W.B. Complete life cycle of the lichen fungus Calopadia puiggarii (Pilocarpaceae, Ascomycetes) documented in situ: Propagule dispersal, establishment of symbiosis, thallus development, and formation of sexual and asexual reproductive structures. Am. J. Bot. 2014, 101, 1836–1848. [Google Scholar] [CrossRef]
- Zhou, J.; Lyu, Y.H.; Richlen, M.L.; Anderson, D.M.; Cai, Z.H. Quorum sensing is a language of chemical signals and plays an ecological role in algal-bacterial interaction. Crit. Rev. Plant Sci. 2016, 1, 82–105. [Google Scholar] [CrossRef]
- Zheng, T.L. (Ed.) The Microbial Control of Harmful Algal Bloom; Xiamen University: Xiamen, China, 2011. [Google Scholar]
- Taylor, J.D.; Cottingham, S.D.; Billinge, J.; Cunliffe, M. Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J. 2014, 8, 245–248. [Google Scholar] [CrossRef]
- Needham, D.M.; Fuhrman, J.A. Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat Microbiol. 2016, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses d-major players in the global ecosystems. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Suttle, C.A.; Chan, A.M.; Cottrell, M.T. Infection of phytoplankton by viruses and reduction of primary productivity. Nature 1990, 347, 467–469. [Google Scholar] [CrossRef]
- Proctor, L.M.; Fuhrman, J.A. Viral mortality of marine bacteria and cyanobacteria. Nature 1990, 343, 60–62. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Van Hannen, E.J.; Zwart, G.; van Agterveld, M.P.; Gons, H.J.; Ebert, J.; Laanbroek, H.J. Changes in bacterial and eukaryotic community structure after mass lysis of filamentous cyanobacteria associated with viruses. Appl. Environ. Microbiol. 1999, 65, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Calbet, A.; Alcaraz, M.; Atienza, D.; Broglio, E.; Vaquè, D. Zooplankton biomass distribution patterns along the western Antarctic Peninsula (December 2002). J. Plankton Res. 2005, 27, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.M.; Quetin, L.B.; Martinson, D.G.; Iannuzzi, R.A.; Stammerjohn, S.E.; Smith, R.C. Palmer LTER: Patterns of distribution of five dominant zooplankton species in the epipelagic zone west of the Antarctic Peninsula, 1993–2004. Deep-Sea Res. Part II 2008, 55, 086–2105. [Google Scholar] [CrossRef]
- Evans, C.; Brussaard, C.P. Regional variation in lytic and lysogenic viral infection in the Southern Ocean and its contribution to biogeochemical cycling. Appl. Environ. Microbiol. 2012, 78, 6741–6748. [Google Scholar] [CrossRef] [Green Version]
- Tomaru, Y.; Toyoda, K.; Kimura, K.; Hata, N.; Yoshida, M.; Nagasaki, K. First evidence for the existence of pennate diatom viruses. ISME J. 2012, 6, 1445–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaru, Y.; Toyoda, K.; Kimura, K.; Takao, Y.; Sakurada, K.; Nakayama, N.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus that infects the marine planktonic diatom Chaetoceros sp. (SS08-C03). Phycol. Res. 2013, 61, 27–36. [Google Scholar] [CrossRef]
- Tomaru, Y.; Toyoda, K.; Suzuki, H.; Nagumo, T.; Kimura, K.; Takao, Y. New single-stranded DNA virus with a unique genomic structure that infects marine diatom Chaetoceros setoensis. Sci. Rep. 2013, 3, 3337. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.H.; Rose, J.B.; Jiang, S.C.; Kellogg, C.A.; Dickson, L. Distribution of viral abundance in the reef environment of Key Largo, Florida. Appl. Environ. Microbiol. 1993, 59, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Weinbauer, M.G.; Suttle, C.A. Comparison of epifluorescence and transmission electron microscopy for counting viruses in natural marine waters. Aqua. Microbial Ecol. 1997, 13, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Clasen, J.L.; Brigden, S.M.; Payet, J.P.; Suttle, C.A. Evidence that viral abundance across oceans and lakes is driven by different biological factors. Freshw. Biol. 2008, 53, 1090–1100. [Google Scholar] [CrossRef]
- Castberg, T.; Larsen, A.; Sandaa, R.A.; Brussaard, C.P.D.; Egge, J.K.; Heldal, M.; Thyrhaug, R.; van Hannen, E.J.; Brathak, G. Microbial population dynamics and diversity during abloom of the marine coccolithophorid Emiliania huxleyi (haptophyta). Mar. Ecol. Prog. Ser. 2001, 221, 39–46. [Google Scholar] [CrossRef]
- Larsen, A.; Castberg, T.; Sandaa, R.A.; Brussaard, C.P.D.; Egge, J.; Heldal, M.; Paulino, A.; Thyrhaug, R.; van Hannen, E.J.; Bratbak, G. Population dynamics and diversity of phytoplankton, bacteria and viruses in a seawater enclosure. Mar. Ecol. Prog. Ser. 2001, 221, 47–57. [Google Scholar] [CrossRef]
- Martínez, J.M.; Schroeder, D.C.; Wilson, W.H. Dynamics and genotypic composition of Emiliania huxleyi and their co-occurring viruses during a Coccolithophore bloom in the North Sea. FEMS Microbiol. Ecol. 2012, 81, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Garretto, A.; Hatzopoulos, T.; Putonti, C. virMine: Automated detection of viral sequences from complex metagenomic samples. Peer J. 2019, 7, e6695. [Google Scholar] [CrossRef]
- Murphy, J.; Riley, J.P. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 1962, 27, 30. [Google Scholar] [CrossRef]
- Greenberg, A.E.; Clesceri, L.S.; Eaton, A.D. Standard Methods for the Examination of Water and Wastewater; American Public Health Association: Washington, DC, USA, 1992. [Google Scholar]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Schluter, D. A variance test for detecting species associations, with some exampleapplications. Ecology 1984, 65, 998–1005. [Google Scholar] [CrossRef]
- Sun, J.Y.; Song, Y.; Ma, Z.P.; Zhang, H.J.; Yang, Z.D.; Cai, Z.H.; Zhou, J. Fungal community dynamics during a marine dinoflagellate (Noctiluca scintillans) bloom. Mar. Environ. Res. 2017, 131, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jiang, X.; Chai, B.; Ma, L.; Li, B.; Zhang, A.; Cole, J.R.; Tiedje, J.M.; Zhang, T. ARGs-OAP: Online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics 2016, 32, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm andits applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 45–477. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource forautomated carbohydrate-active enzyme annotation. Nucleic. Acids. Res. 2012, 40, 445–451. [Google Scholar] [CrossRef]
- Huang, X.Q.; Zhu, J.M.; Cai, Z.H.; Lao, Y.M.; Jin, H.; Yu, K.; Zhang, B.Y.; Zhou, J. Profiles of quorum sensing (QS)-related sequences in phycospheric microorganisms during a marine dinoflagellate bloom, as determined by a metagenomic approach. Microbiol. Res. 2018, 217, 1–13. [Google Scholar] [CrossRef]
- Zheng, T.L.; Li, W.; Li, Y. Advance in study on microbial control of harmful algae blooms-exploitation and research on marine algicidal bacteria. J. Xiamen Univ. 2011, 10, 1658–1668. [Google Scholar]
- Yang, Q.W.; Gao, C.; Jian, Y.; Wang, M.; Zhou, X.H.; Shao, H.B.; Gong, Z.; McMinn, A. Metagenomic characterization of the viral community of the South Scotia Ridge. Viruses 2019, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Liang, Y.T.; Wang, M.; Liang, Y.T.; Wang, M.; Jiang, Y.; Yang, Q.W.; Xia, J. Viral diversity and its relationship with environmental factors at the surface and deep sea of Prydz Bay, Antarctica. Front. Microbiol. 2018, 9, 2981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Wang, K.; Shen, L.X.; Chen, H.P.; Hou, F.R.; Zhou, X.Y.; Zhang, D.M.; Zhu, X.Y. Microbial community dynamics and assembly follow trajectories of an early-spring diatom bloom in a semi-enclosed bay. Appl. Environ. Microbiol. 2018, 84, e01000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Park, S.Y.; Park, M.; Lee, S.; Lee, T.K. Seasonal dynamics and metagenomic characterization of marine viruses in Goseong Bay, Korea. PLoS ONE 2017, 12, e0169841. [Google Scholar] [CrossRef] [PubMed]
- Kallies, R.; Hölzer, M.; Toscan, R.B.; da Rocha, U.N.; Anders, J.; Marz, M.; Chatzinotas, A. Evaluation of sequencing library preparation protocols for viral metagenomic analysis from pristine aquifer ground waters. Viruses 2019, 11, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danovaro, R.; Corinaldesi, C.; Dell’Anno, A.; Fabiano, M.; Corselli, C. Viruses, prokaryotes and DNA in the sediments of a deep-hypersaline anoxic basin (DHAB) of the Mediterranean Sea. Environ. Microbiol. 2005, 7, 586–592. [Google Scholar] [CrossRef]
- Borin, S.; Crotti, E.; Mapelli, F.; Tamagnini, I.; Corselli, C.; Daffonchio, D. DNA is preserved and maintains transforming potential after contact with brines of the deep anoxic hypersaline lakes of the Eastern Mediterranean Sea. Saline Syst. 2008, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Corinaldesi, C.; Tangherlini, M.; Luna, G.M.; Dell’anno, A. Extracellular DNA can preserve the genetic signatures of present and past viral infection events in deep hypersaline anoxic basins. Proc. Biol. Sci. 2014, 281, 20133299. [Google Scholar] [CrossRef]
- Antunes, A.; Alam, I.; Simoes, M.F.; Daniels, C.; Ferreira, A.J.S.; Siam, R.; El-Dorry, H.; Bajic, V.B. First insights into the viral communities of the Deep-Sea anoxic brines of the Red Sea. Genom. Proteom. Bioinf. 2015, 13, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef]
- Kim, K.H.; Chang, H.W.; Nam, Y.D.; Roh, S.W.; Kim, M.S.; Sung, Y.; Jeon, C.O.; Oh, H.M.; Bae, J.W. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon-Schumacher, T.; Guajardo-Leiva, S.; Anton, J.; Diez, B. Elucidating viral communities during a phytoplankton bloom on the West Antarctic Peninsula. Front. Microbiol. 2018, 10, 1014. [Google Scholar] [CrossRef]
- Deng, J.; Qin, B.; Paerl, H.W.; Zhang, Y.; Wu, P.; Ma, J.; Chen, Y. Effects of nutrients, temperature and their interactions on spring phytoplankton community succession in lake Taihu, China. PLoS ONE 2014, 9, e113960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Kloclow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.X.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Zhou, J.; Zhu, X.S.; Yu, S.C.; Cai, Z.H. An association network analysis among microeukaryotes and bacterioplankton reveals algal bloom dynamics. J. Phycol. 2015, 51, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Luo, E.; Aylward, F.O.; Mende, D.R.; DeLong, E.F. Bacteriophage distributions and temporal variability in the ocean’s interior. mBio 2017, 8, e01903-17. [Google Scholar] [CrossRef] [Green Version]
- Alberti, A.; Poulain, J.; Engelen, S.; Labadie, K.; Romac, S.; Ferrera, I.; Albini, G.; Aury, J.M.; Belser, C.; Bertrand, A.; et al. Viral to metazoan marine plankton nucleotide sequences from the Tara Oceans expedition. Sci. Data 2017, 4, 170093. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, T.V.; Larsen, A.; Bratbak, G.; Pagarete, A.; Edvardsen, B.; Egge, E.D.; Sandaa, R.A. Seasonal dynamics of haptophytes and dsDNA algal viruses suggest complex virus-host relationship. Viruses 2017, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Klumpp, J.; Fouts, D.E.; Sozhamannan, S. Bacteriophage functional genomics and its role in bacterial pathogen detection. Brief. Func. Genom. 2013, 12, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, B.L.; Westveld, A.H.; Brum, J.R.; Sullivan, M.B. Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proc. Natl. Acad. Sci. USA 2014, 111, 10714–10719. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Jin, M.; Li, L.; Qu, W.; Zeng, R. Genome sequencing reveals the complex polysaccharide-degrading ability of novel deep-sea bacterium Flammeovirga pacifica WPAGA1. Front. Microbiol. 2017, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Emerson, J.B.; Roux, S.; Brum, J.R.; Bolduc, B.; Woodcroft, B.J.; Jang, H.B.; Singleton, C.M.; Solden, L.M.; Naas, A.E.; Boyd, J.A.; et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 2018, 3, 870–880. [Google Scholar] [CrossRef]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, B.L.; U’Ren, J.M. Viral metabolic reprogramming in marine ecosystems. Curr. Opin. Microbiol. 2016, 31, 161–168. [Google Scholar] [CrossRef]
- Maaroufi, H.; Levesque, R.C. Glycoside hydrolase family 32 is present in Bacillus subtilis phages. Virol. J. 2015, 12, 157. [Google Scholar] [CrossRef] [Green Version]
- Davison, M.; Treangen, T.J.; Koren, S.; Pop, M.; Bhaya, D. Diversity in a polymicrobial community revealed by analysis of viromes, endolysins and CRISPR spacers. PLoS ONE 2016, 11, e0160574. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.L.; Sullivan, M.B.; Fernando, S.C. Dietary energy drives the dynamic response of bovine rumen viral communities. Microbiome 2017, 5, 155. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.-P.; Cai, Z.-H.; Zuo, P.; Meng, F.-X.; Zhu, J.-M.; Zhou, J. Temporal Variability of Virioplankton during a Gymnodinium catenatum Algal Bloom. Microorganisms 2020, 8, 107. https://doi.org/10.3390/microorganisms8010107
Du X-P, Cai Z-H, Zuo P, Meng F-X, Zhu J-M, Zhou J. Temporal Variability of Virioplankton during a Gymnodinium catenatum Algal Bloom. Microorganisms. 2020; 8(1):107. https://doi.org/10.3390/microorganisms8010107
Chicago/Turabian StyleDu, Xiao-Peng, Zhong-Hua Cai, Ping Zuo, Fan-Xu Meng, Jian-Ming Zhu, and Jin Zhou. 2020. "Temporal Variability of Virioplankton during a Gymnodinium catenatum Algal Bloom" Microorganisms 8, no. 1: 107. https://doi.org/10.3390/microorganisms8010107
APA StyleDu, X.-P., Cai, Z.-H., Zuo, P., Meng, F.-X., Zhu, J.-M., & Zhou, J. (2020). Temporal Variability of Virioplankton during a Gymnodinium catenatum Algal Bloom. Microorganisms, 8(1), 107. https://doi.org/10.3390/microorganisms8010107