HybridMine: A Pipeline for Allele Inheritance and Gene Copy Number Prediction in Hybrid Genomes and Its Application to Industrial Yeasts


Abstract
:1. Introduction
2. Materials and Methods
2.1. HybridMine Pipeline Architecture
2.2. Validation Dataset
2.3. Statistical Analysis
2.4. Homologs Validation
2.5. Benchmark Validation of HybridMine
2.6. Code Availability
2.7. Data Availability
3. Results
3.1. Allele Inheritance Prediction Pipeline
3.2. HybridMine Usage
3.3. Paralogs Validation
3.4. Benchmark Evaluation
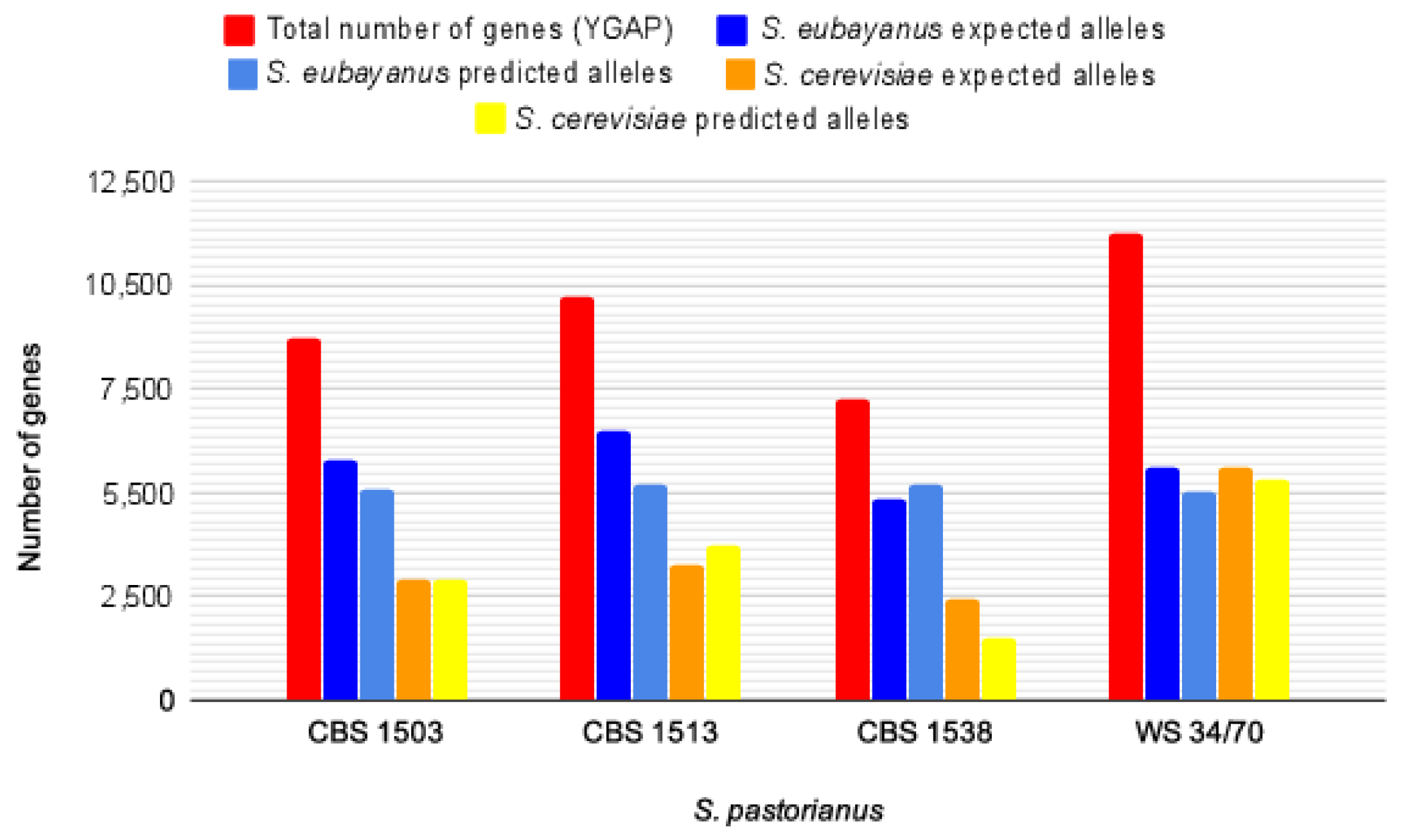
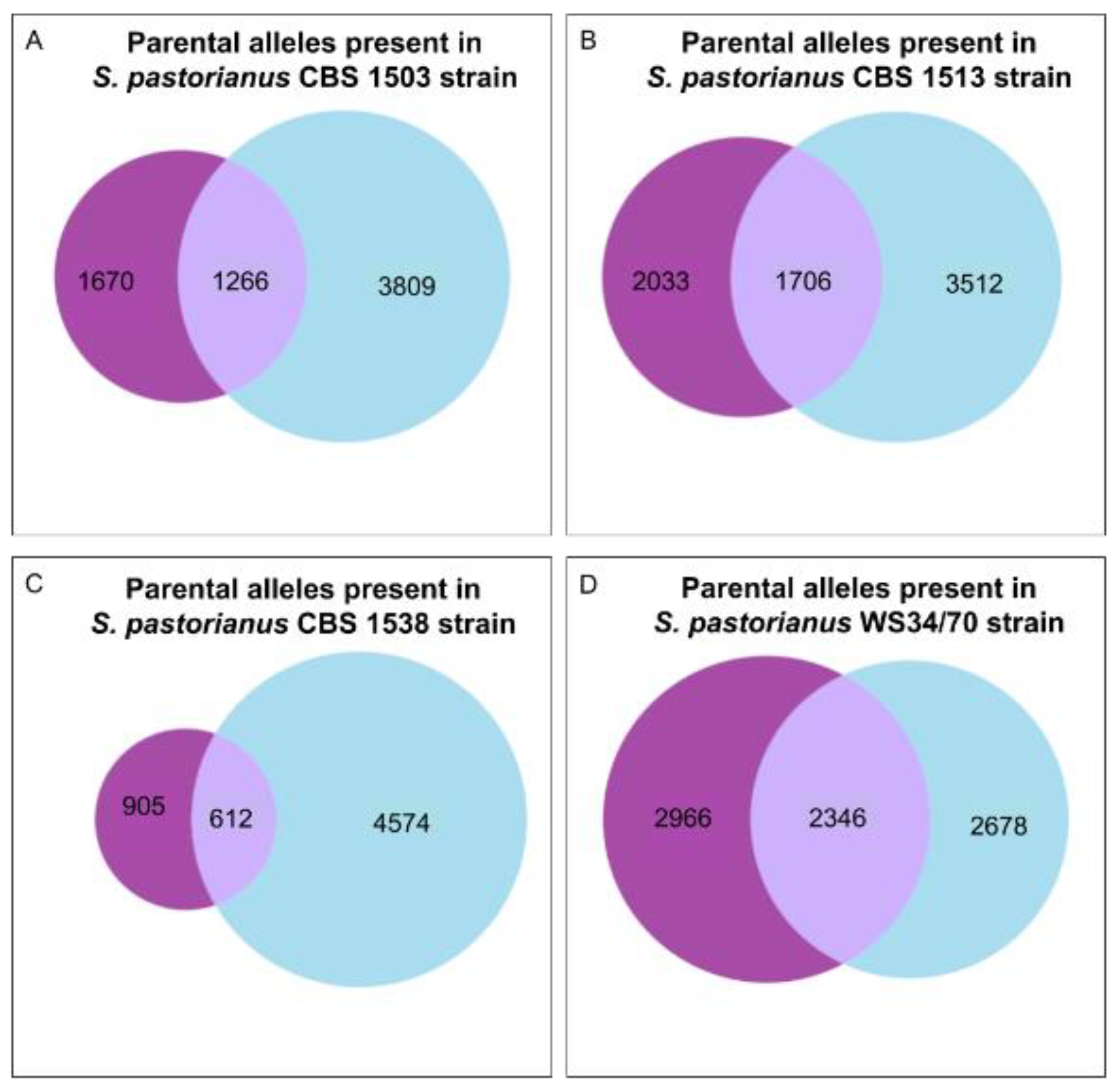
3.5. Application of HybridMine to Annotate S. pastorianus Hybrids and Validation of Outputs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, W.; Averette, A.F.; Desnos-Ollivier, M.; Ni, M.; Dromer, F.; Heitman, J. Genetic Diversity and Genomic Plasticity of Cryptococcus neoformans AD Hybrid Strains. G3 Genes Genomes Genet. 2012, 2, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef]
- Monerawela, C.; Bond, U. Brewing up a storm: The genomes of lager yeasts and how they evolved. Biotechnol. Adv. 2017, 35, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Piatkowska, E.M.; Naseeb, S.; Knight, D.; Delneri, D. Chimeric protein complexes in hybrid species generate novel phenotypes. PLoS Genet. 2013, 9, e1003836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, B.; Sherlock, G. Reconstruction of the genome origins and evolution of the hybrid lager yeast saccharomyces pastorianus. Genome Res. 2008, 18, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Liti, G.; Peruffo, A.; James, S.A.; Roberts, I.N.; Louis, E.J. Inferences of evolutionary relationships from a population survey of ltr-retrotransposons and telomeric-associated sequences in the saccharomyces sensu stricto complex. Yeast 2005, 22, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Monerawela, C.; James, T.C.; Wolfe, K.H.; Bond, U. Loss of lager specific genes and subtelomeric regions define two different saccharomyces cerevisiae lineages for saccharomyces pastorianus group i and ii strains. FEMS Yeast Res. 2015, 15, fou008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsammar, H.; Delneri, D. An update on the diversity, ecology and biogeography of the Saccharomyces genus. FEMS Yeast Res. 2020, 20, foaa013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, U.; Neal, C.; Donnelly, D.; James, T.C. Aneuploidy and copy number breakpoints in the genome of lager yeasts mapped by microarray hybridisation. Curr. Genet. 2004, 45, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Noonan, G.J. New Brewing Lager Beer: The Most Comprehensive Book for Home- and Microbrewers; Brewers Publications: Boulder, CO, USA, 1996. [Google Scholar]
- Bing, J.; Han, P.-J.; Liu, W.-Q.; Wang, Q.-M.; Bai, F.-Y. Evidence for a Far East Asian origin of lager beer yeast. Curr. Biol. 2014, 24, R380–R381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan-Martini, A.; Martini, A. A Taxonomic Key for the Genus Saccharomyces. Syst. Appl. Microbiol. 1993, 16, 113–119. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H. Origin of the yeast whole-genome duplication. PLoS Biol. 2015, 13, e1002221. [Google Scholar] [CrossRef] [Green Version]
- Katju, V.; Farslow, J.C.; Bergthorsson, U. Variation in gene duplicates with low synonymous divergence in Saccharomyces cerevisiae relative to Caenorhabditis elegans. Genome Biol. 2009, 10, R75. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.; Liti, G. Saccharomyces pastorianus: Genomic insights inspiring innovation for industry. Yeast 2014, 32, 17–27. [Google Scholar] [CrossRef]
- Bloomberg Business, Beer Processing Market Worth $815.4 Billion by 2025; MarketsandMarkets™; MarketsandMarkets: Chicago, IL, USA, 2019.
- Hewitt, S.; Donaldson, I.; Lovell, S.C.; Delneri, D. Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS ONE 2014, 9, e92203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, A.; Hesselbart, A.; Wendland, J. Genome Sequence of Saccharomyces carlsbergensis, the World’s First Pure Culture Lager Yeast. G3 Genes Genomes Genet. 2014, 4, 783–793. [Google Scholar]
- Okuno, M.; Kajitani, R.; Ryusui, R.; Morimoto, H.; Kodama, Y.; Itoh, T. Next-generation sequencing analysis of lager brewing yeast strains reveals the evolutionary history of interspecies hybridization. DNA Res. 2016, 23, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Goetz, S.; García-Gómez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Proux-Wéra, E.; Armisen, D.; Byrne, K.P.; Wolfe, K.H. A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach. BMC Bioinform. 2012, 13, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinty, R.J.; Rubinstein, R.G.; Neil, A.J.; Dominska, M.; Kiktev, D.; Petes, T.D.; Mirkin, S.M. Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair. Genome Res. 2017, 27, 2072–2082. [Google Scholar] [CrossRef] [Green Version]
- Langdon, Q.K.; Peris, D.; Kyle, B.; Hittinger, C.T. sppIDer: A Species Identification Tool to Investigate Hybrid Genomes with High-Throughput Sequencing. Mol. Biol. Evol. 2018, 35, 2835–2849. [Google Scholar] [CrossRef] [Green Version]
- Tattini, L.; Tellini, N.; Mozzachiodi, S.; D’Angiolo, M.; Loeillet, S.; Nicolas, A.; Liti, G. Accurate Tracking of the Mutational Landscape of Diploid Hybrid Genomes. Mol. Biol. Evol. 2019, 36, 2861–2877. [Google Scholar] [CrossRef] [Green Version]
- De León-Medina, P.M.; Elizondo-Gonzalez, R.; Damas-Buenrostro, L.C.; Geertman, J.-M.; Broek, M.V.D.; Galan-Wong, L.J.; Ortiz-Lopez, R.; Pereyra-Alférez, B. Genome annotation of a Saccharomyces sp. lager brewer’s yeast. Genom. Data 2016, 9, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Broek, M.V.D.; Bolat, I.; Nijkamp, J.F.; Ramos, E.; Luttik, M.A.H.; Koopman, F.; Geertman, J.M.; De Ridder, D.; Pronk, J.; Daran, J.-M. Chromosomal Copy Number Variation in Saccharomyces pastorianus Is Evidence for Extensive Genome Dynamics in Industrial Lager Brewing Strains. Appl. Environ. Microbiol. 2015, 81, 6253–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, Y.; Kanamori, T.; Itoh, T.; Kodama, Y.; Rainieri, S.; Nakamura, N.; Shimonaga, T.; Hattori, M.; Ashikari, T. Genome sequence of the lager brewing yeast, an interspecies hybrid. DNA Res. 2009, 16, 115–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parental Genomes | Prediction | eggNOG-Mapper | Blast2GO | HybridMine | |
|---|---|---|---|---|---|
| S. pastorianus WS34/70 | S. cerevisiae | Homologs | NA | 5373 | 8182 |
| 1:1 Orthologs | 9997 | NA | 5312 | ||
| Paralogs | NA | NA | 1738 | ||
| S. eubayanus | Homologs | NA | 6148 | 8222 | |
| 1:1 Orthologs | 0 | NA | 5024 | ||
| Paralogs | NA | NA | 1586 | ||
| Search only wide taxa group | Search specific to the parental genomes | Search specific to the parental genomes | |||
| Free tool | Tool under license | Free tool | |||
| S. pastorianus Strain Annotated in This Study | CBS 1538 | CBS 1503 | CBS 1513 | WS 34/70 |
|---|---|---|---|---|
| Total Number of Genes | 7288 | 8714 | 9728 | 11,265 |
| Number of Predicted Alleles in S. eubayanus | 5186 | 5075 | 5218 | 5024 |
| Number of Predicted Alleles in S. cerevisiae | 1517 | 2936 | 3739 | 5312 |
| 1:1 Orthologs with Same Sequence Identity in Both Parents | 9 | 10 | 9 | 7 |
| ORFs with less than 80% Sequence Identity With a Predicted Parental Allele | 9 | 3 | 4 | 2 |
| S. pastorianus Genes With no Parental Allele Predicted | 567 | 690 | 758 | 920 |
| Number of Different Paralogs | 2814 | 3056 | 3316 | 3652 |
| S. pastorianus WS 34/70 Strain Genes | Best Homolog in S. cerevisiae | Best Homolog in S. eubayanus | Parental Allele Prediction with HybridMine | Parental Allele Phylogenetic Tree Output |
|---|---|---|---|---|
| SPGP0N03370 | YOL158C | XP_018222854.1 | YOL158C | YOL158C |
| SPGP0I00140 | YBR296C | XP_018223320.1 | XP_018223320.1 | XP_018223320.1 |
| SPGP0I00180 | YBR290W | XP_018223316.1 | XP_018223316.1 | XP_018223316.1 |
| SPGP0J00870 | YBR215W | XP_018223244.1 | YBR215W | YBR215W |
| SPGP0J00840 | YBR218C | XP_018223247.1 | YBR218C | YBR218C |
| SPGP0R01550 | YGL062W | XP_018222170.1 | YGL062W | YGL062W |
| SPGP0I00870 | YBR218C | XP_018223247.1 | XP_018223247.1 | XP_018223247.1 |
| SPGP0E03270 | YGL062W | XP_018222170.1 | XP_018222170.1 | XP_018222170.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timouma, S.; Schwartz, J.-M.; Delneri, D. HybridMine: A Pipeline for Allele Inheritance and Gene Copy Number Prediction in Hybrid Genomes and Its Application to Industrial Yeasts. Microorganisms 2020, 8, 1554. https://doi.org/10.3390/microorganisms8101554
Timouma S, Schwartz J-M, Delneri D. HybridMine: A Pipeline for Allele Inheritance and Gene Copy Number Prediction in Hybrid Genomes and Its Application to Industrial Yeasts. Microorganisms. 2020; 8(10):1554. https://doi.org/10.3390/microorganisms8101554
Chicago/Turabian StyleTimouma, Soukaina, Jean-Marc Schwartz, and Daniela Delneri. 2020. "HybridMine: A Pipeline for Allele Inheritance and Gene Copy Number Prediction in Hybrid Genomes and Its Application to Industrial Yeasts" Microorganisms 8, no. 10: 1554. https://doi.org/10.3390/microorganisms8101554