Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis


Abstract
:1. Introduction
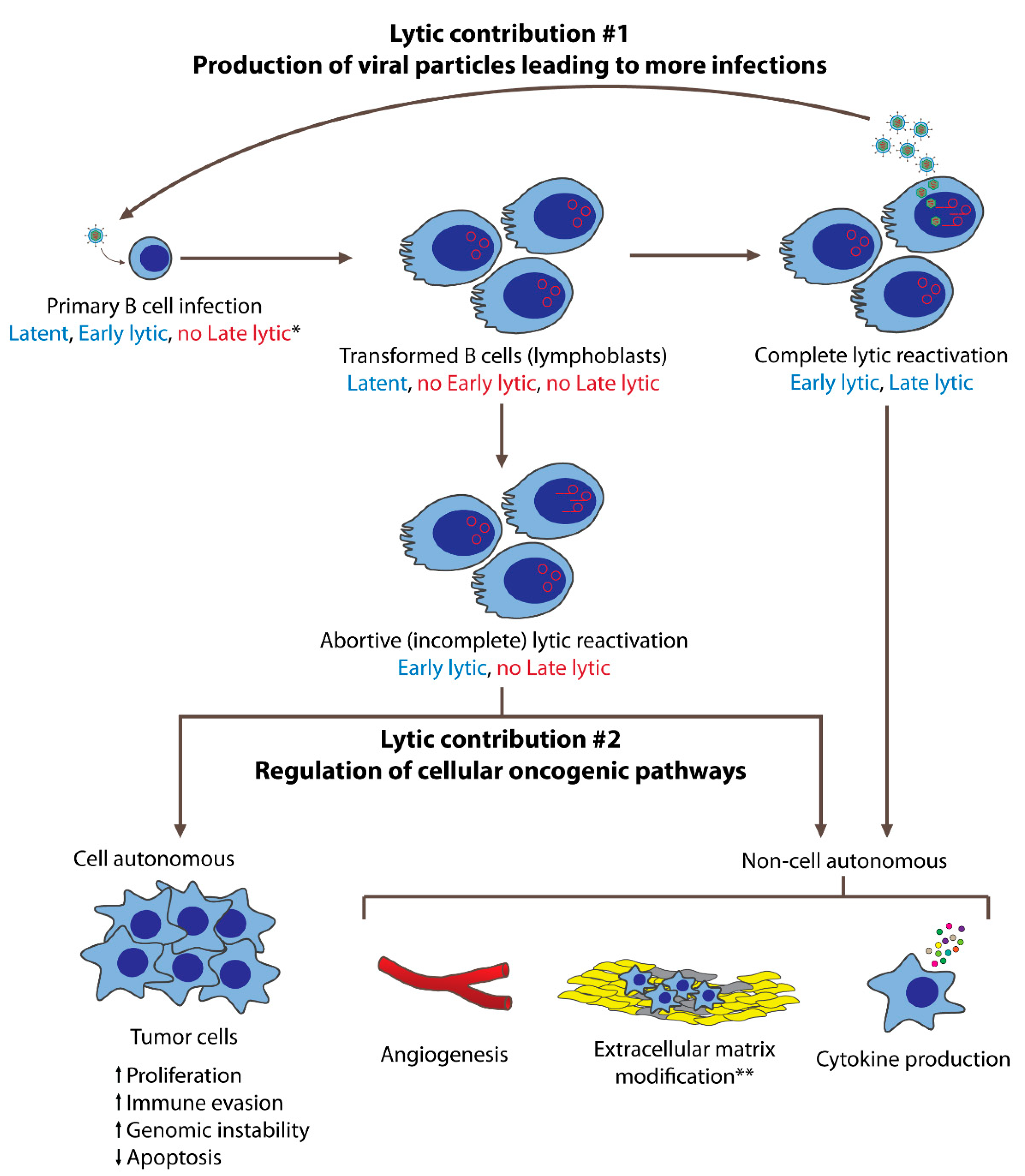
2. EBV’s Lytic Phase Contributes to Tumorigenesis by Production of Infectious Viral Particles
3. EBV’s Lytic Gene Expression in EBV-Associated Tumor Samples
4. Functional Contributions of EBV’s Lytic Genes to EBV’s Oncogenesis
5. Cellular Regulation of Tumorigenesis by EBV’s Lytic Phase
5.1. Immunomodulation and Immune Evasion
5.2. Angiogenesis and Invasion
5.3. Genomic Instability
5.4. Cell Cycle Regulation and Apoptosis
6. EBV’s Lytic miRNAs in Tumorigenesis
7. Inhibitor Studies: A Test for a Role for EBV’s Lytic Phase in Oncogenesis?
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sánchez, A.; Fuentes-Panana, E.M. The immunomodulatory capacity of an epstein-barr virus abortive lytic cycle: Potential contribution to viral tumorigenesis. Cancers 2018, 10, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; Bode, A.M.; Cao, Y. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int. J. Biol. Sci. 2016, 12, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Tumor Microenvironment Conditioning by Abortive Lytic Replication of Oncogenic γ-Herpesviruses. Adv. Exp. Med. Biol. 2020, 1225, 127–135. [Google Scholar] [CrossRef]
- Lwoff, A. Lysogeny. Bacteriol. Rev. 1953, 17, 269–337. [Google Scholar] [CrossRef]
- Sugden, B.; Phelps, M.; Domoradzki, J. Epstein-Barr virus DNA is amplified in transformed lymphocytes. J. Virol. 1979, 31, 590–595. [Google Scholar] [CrossRef] [Green Version]
- Honess, R.W.; Roizman, B. Regulation of Herpesvirus Macromolecular Synthesis I. Cascade Regulation of the Synthesis of Three Groups of Viral Proteins 1. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.K. Correlation between HSV-1 DNA sequence and viral transcription maps. In Herpesvirus Transcription and Its Regulation; Wagner, E.K., Ed.; CRC-Press: Boca Raton, FL, USA, 1991; pp. 29–47. [Google Scholar]
- Jochum, S.; Ruiss, R.; Moosmann, A.; Hammerschmidt, W.; Zeidler, R. RNAs in Epstein-Barr virions control early steps of infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1396–E1404. [Google Scholar] [CrossRef] [Green Version]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First Days in the Life of Naive Human B Lymphocytes Infected with Epstein-Barr Virus. mBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr Virus BZLF1 Gene, a Switch from Latency to Lytic Infection, Is Expressed as an Immediate-Early Gene after Primary Infection of B Lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med. Virol. 2014, 24, 57242–57253. [Google Scholar] [CrossRef] [PubMed]
- De-Thé, G.; Geser, A.; Day, N.E.; Tukei, P.M.; Williams, E.M.; Beri, D.P.; Smith, P.G.; Dean, A.G.; Bornkamm, G.W.; Feorino, P.; et al. Epidemiological evidence for causal relationship between Epstein-Barr virus and Burkitt’s lymphoma from Ugandan prospective study. Nature 1978, 274, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Geser, A.; De Thé, G.; Lenoir, G.; Day, N.E.; Williams, E.H. Final case reporting from the ugandan prospective study of the relationship between ebv and burktit’s lymphoma. Int. J. Cancer 1982, 29, 397–400. [Google Scholar] [CrossRef]
- Burkitt, D.P. Etiology of Burkitt’s Lymphoma—An Alternative Hypothesis to a Vectored Virus 1. J. Natl. Cancer Inst. Natl. Cancer Inst. 1969, 42, 19–28. [Google Scholar]
- Whittle, H.C.; Brown, J.; Marsh, K.; Greenwood, B.M.; Seidelin, P.; Tighe, H.; Wedderburn, L. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature 1984, 312, 449–450. [Google Scholar] [CrossRef]
- Lam, K.M.C.; Syed, N.; Crawford, D.H.; Whittle, H. Circulating Epstein-Barr virus-carrying B cells in acute malaria. Lancet 1991, 337, 876–878. [Google Scholar] [CrossRef]
- Manolov, G.; Manolova, Y. Marker band in one chromosome 14 from burkitt lymphomas. Nature 1972, 237, 33–34. [Google Scholar] [CrossRef]
- Taub, R.; Moulding, C.; Battey, J.; Murphy, W.; Vasicek, T.; Lenoir, G.M.; Leder, P. Activation and somatic mutation of the translocated c-myc gene in Burkitt lymphoma cells. Cell 1984, 36, 339–348. [Google Scholar] [CrossRef]
- Rowley, J.D. Identification of the constant chromosome regions involved in human hematologic malignant disease. Science 1982, 216, 749–751. [Google Scholar] [CrossRef]
- Hayday, A.C.; Gillies, S.D.; Saito, H.; Wood, C.; Wiman, K.; Hayward, W.S.; Tonegawa, S. Activation of a translocated human c-myc gene by an enhancer in the immunoglobulin heavy-chain locus. Nature 1984, 307, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.; Lipman, M. Comparison of the yield of infectious virus from clones of human and simian lymphoblastoid lines transformed by epstein-barr virus. J. Exp. Med. 1973, 138, 1398–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornkamm, G.W.; Delius, H.; Zimber, U.; Hudewentz, J.; Epstein, M.A. Comparison of Epstein-Barr virus strains of different origin by analysis of the viral DNAs. J. Virol. 1980, 35, 603–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gärtner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous Lytic Replication and Epitheliotropism Define an Epstein-Barr Virus Strain Found in Carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Delecluse, S.; Poirey, R.; Zeier, M.; Schnitzler, P.; Behrends, U.; Tsai, M.-H.; Delecluse, H.-J. Identification and Cloning of a New Western Epstein-Barr Virus Strain that Efficiently Replicates in Primary B Cells. J. Virol. 2020, 94, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yao, Q.Y.; Rooney, C.M.; Sculley, T.B.; Moss, D.J.; Rupani, H.; Laux, G.; Bornkamm, G.W.; Rickinson, A.B. New type B isolates of Epstein-Barr virus from Burkitt’s lymphoma and from normal individuals in endemic areas. J. Gen. Virol. 1987, 68 Pt 11, 2853–2862. [Google Scholar] [CrossRef]
- Yu, X.; McCarthy, P.J.; Wang, Z.; Gorlen, D.A.; Mertz, J.E. Shutoff of BZLF1 Gene Expression Is Necessary for Immortalization of Primary B Cells by Epstein-Barr Virus. J. Virol. 2012, 86, 8086–8096. [Google Scholar] [CrossRef] [Green Version]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef] [Green Version]
- Romero-Masters, J.C.; Huebner, S.M.; Ohashi, M.; Bristol, J.A.; Benner, B.E.; Barlow, E.A.; Turk, G.L.; Nelson, S.E.; Baiu, D.C.; van Sciver, N.; et al. B cells infected with Type 2 Epstein-Barr virus (EBV) have increased NFATc1/NFATc2 activity and enhanced lytic gene expression in comparison to Type 1 EBV infection. PLoS Pathog. 2020, 16, e1008365. [Google Scholar] [CrossRef] [Green Version]
- Sugden, B. Expression of virus-associated functions in cells trnasformed in vitro by Epstein-Barr virus: Epstein-Barr virus cell surface antigen and virus-release from transformed cells. In Immune Deficiency and Cancer: Epstein-Barr Virus and Lymphoproliferative Malignancies; Purtilo, D.T., Ed.; Plenum Publishing Corporation: New York, NY, USA, 1984; pp. 165–177. [Google Scholar]
- Kintner, C.; Sugden, B. Conservation and progressive methylation of Epstein-Barr viral DNA sequences in transformed cells. J. Virol. 1981, 38, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Bhende, P.M.; Seaman, W.T.; Delecluse, H.J.; Kenney, S.C. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 2004, 36, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, M.; Gobel, C.; Hammerschmidt, W. The Lytic Phase of Epstein-Barr Virus Requires a Viral Genome with 5-Methylcytosine Residues in CpG Sites. J. Virol. 2012, 86, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef]
- Cochet, C.; Martel-Renoir, D.; Grunewald, V.; Bosq, J.; Cochet, G.; Schwaab, G.; Bernaudin, J.-F.; Joab, I. Expression of the Epstein-Barr Virus Immediate Early Gen, BZLF1, in Nasopharyngeal Carcinoma Tumor Cells. Virology 1993, 197, 358–365. [Google Scholar] [CrossRef]
- Ramayanti, O.; Juwana, H.; Verkuijlen, S.A.M.W.; Adham, M.; Pegtel, M.D.; Greijer, A.E.; Middeldorp, J.M. Epstein-Barr virus mRNA profiles and viral DNA methylation status in nasopharyngeal brushings from nasopharyngeal carcinoma patients reflect tumor origin. Int. J. Cancer 2017, 140, 149–162. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr Virus Genomes and Expression Profiles in Gastric Adenocarcinoma. J. Virol. 2017, 92, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Djavadian, R.; Chiu, Y.F.; Johannsen, E. An Epstein-Barr Virus-Encoded Protein Complex Requires an Origin of Lytic Replication In Cis to Mediate Late Gene Transcription. PLoS Pathog. 2016, 12, e1005718. [Google Scholar] [CrossRef] [Green Version]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 1992, 66, 5030–5039. [Google Scholar] [CrossRef] [Green Version]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Replication of Epstein-Barr virus oriLyt: Lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 1995, 69, 2998–3006. [Google Scholar] [CrossRef] [Green Version]
- Djavadian, R.; Hayes, M.; Johannsen, E. CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 Kinetic classes from 2 mechanisms. PLoS Pathog. 2018, 14, e1007114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.-H.; Delecluse, H.-J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr Virus Lytic Infection Contributes to Lymphoproliferative Disease in a SCID Mouse Model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.-D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A New Model of Epstein-Barr Virus Infection Reveals an Important Role for Early Lytic Viral Protein Expression in the Development of Lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Burns, D.M.; Tierney, R.; Shannon-Lowe, C.; Croudace, J.; Inman, C.; Abbotts, B.; Nagra, S.; Fox, C.P.; Chaganti, S.; Craddock, C.F.; et al. Memory B-cell reconstitution following allogeneic hematopoietic stem cell transplantation is an EBV-associated transformation event. Blood 2015, 126, 2665–2675. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.; Wu, S.-Y.; Chang, S.-S.; Su, I.-J.; Tsai, C.-H.; Lai, S.-J.; Shiau, A.-L.; Takada, K.; Chang, Y. Epstein-Barr Virus Lytic Transactivator Zta Enhances Chemotactic Activity through Induction of Interleukin-8 in Nasopharyngeal Carcinoma Cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Yeh, T.-H.; Lai, H.-C.; Wu, S.-Y.; Su, I.-J.; Takada, K.; Chang, Y. Epstein-Barr Virus Zta-Induced Immunomodulators from Nasopharyngeal Carcinoma Cells Upregulate Interleukin-10 Production from Monocytes. J. Virol. 2011, 85, 7333–7342. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.C.; Lin, S.J.; Chen, P.W.; Luo, W.Y.; Yeh, T.H.; Wang, H.W.; Chen, C.J.; Tsai, C.H. EBV Zta protein induces the expression of interleukin-13, promoting the proliferation of EBV-infected B cells and lymphoblastoid cell lines. Blood 2009, 114, 109–118. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; Van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.H.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.-J.; Hislop, A.D.; Rowe, M. The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M.V. EBV-encoded dUTPase induces immune dysregulation: Implications for the pathophysiology of EBV-associated disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2016, 90, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.W.; Keating, S.; Gomez, R.; Franken, K.L.M.C.; Ottenhoff, T.H.M.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr Virus gp42 Is Posttranslationally Modified to Produce Soluble gp42 that Mediates HLA Class II Immune Evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, T.; Sato, H.; Murono, S.; Pagano, J.S.; Furukawa, M. Matrix metalloproteinase 9 is induced by the Epstein-Barr virus BZLF1 transactivator. Clin. Exp. Metastasis 1999, 17, 431–436. [Google Scholar] [CrossRef]
- Lu, J.; Chua, H.H.; Chen, S.Y.; Chen, J.Y.; Tsai, C.H. Regulation of matrix metalloproteinase-1 by Epstein-Barr virus proteins. Cancer Res. 2003, 63, 256–262. [Google Scholar]
- Lan, Y.Y.; Yeh, T.H.; Lin, W.H.; Wu, S.Y.; Lai, H.C.; Chang, F.H.; Takada, K.; Chang, Y. Epstein-Barr Virus Zta Upregulates Matrix Metalloproteinases 3 and 9 that Synergistically Promote Cell Invasion In Vitro. PLoS ONE 2013, 8, e56121. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.Y.; Chang, F.H.; Tsai, J.H.; Chang, Y. Epstein-Barr virus Rta promotes invasion of bystander tumor cells through paracrine of matrix metalloproteinase 9. Biochem. Biophys. Res. Commun. 2018, 503, 2160–2166. [Google Scholar] [CrossRef]
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [CrossRef]
- Lee, C.-P.; Chen, J.-Y.; Wang, J.-T.; Kimura, K.; Takemoto, A.; Lu, C.-C.; Chen, M.-R. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol. 2007, 81, 5166–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Liu, M.-T.; Chang, Y.-T.; Fang, C.-Y.; Chou, S.-P.; Liao, H.-W.; Kuo, K.-L.; Hsu, S.-L.; Chen, Y.-R.; Wang, P.-W.; et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [Green Version]
- Altmann, M.; Hammerschmidt, W. Epstein-barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An epstein-barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Fitzsimmons, L.; Cartlidge, R.; Chang, C.; Sejic, N.; Galbraith, L.C.A.; Suraweera, C.D.; Croom-Carter, D.; Dewson, G.; Tierney, R.J.; Bell, A.I.; et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020, 27, 1554–1568. [Google Scholar] [CrossRef]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef]
- Schaeffner, M.; Mrozek-Gorska, P.; Buschle, A.; Woellmer, A.; Tagawa, T.; Cernilogar, F.M.; Schotta, G.; Krietenstein, N.; Lieleg, C.; Korber, P.; et al. BZLF1 interacts with chromatin remodelers promoting escape from latent infections with EBV. Life Sci. Alliance 2019, 2, e201800108. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Soto, R.; Damania, B. Modulation of angiogenic processes by the human gammaherpesviruses, Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus. Front. Microbiol. 2019, 10, 1544. [Google Scholar] [CrossRef]
- Hong, G.K.; Kumar, P.; Wang, L.; Damania, B.; Gulley, M.L.; Delecluse, H.-J.; Polverini, P.J.; Kenney, S.C. Epstein-Barr Virus Lytic Infection Is Required for Efficient Production of the Angiogenesis Factor Vascular Endothelial Growth Factor in Lymphoblastoid Cell Lines. J. Virol. 2005, 79, 13984–13992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.J.; Wu, S.W.; Chou, Y.C.; Lin, J.H.; Huang, Y.C.; Chen, M.R.; Ma, N.; Tsai, C.H. Novel expression and regulation of TIMP-1 in Epstein Barr virus-infected cells and its impact on cell survival. Virology 2015, 481, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.Y.; Lee, C.H.; Wu, C.C.; Chang, Y.T.; Yu, S.L.; Chou, S.P.; Huang, P.T.; Chen, C.L.; Hou, J.W.; Chang, Y.; et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int. J. Cancer 2009, 124, 2016–2025. [Google Scholar] [CrossRef]
- Li, R.; Liao, G.; Nirujogi, R.S.; Pinto, S.M.; Shaw, P.G.; Huang, T.C.; Wan, J.; Qian, J.; Gowda, H.; Wu, X.; et al. Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLoS Pathog. 2015, 11, e1005346. [Google Scholar] [CrossRef]
- Bellows, D.S.; Howell, M.; Pearson, C.; Hazlewood, S.A.; Hardwick, J.M. Epstein-Barr Virus BALF1 Is a BCL-2-Like Antagonist of the Herpesvirus Antiapoptotic BCL-2 Proteins. J. Virol. 2002, 76, 2469–2479. [Google Scholar] [CrossRef] [Green Version]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [Green Version]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.S.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus bhrf1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, R.; Fitzsimmons, L.; Thomas, W.A.; Kelly, G.L.; Rowe, M.; Bell, A.I. Quantitative Studies of Epstein-Barr Virus-Encoded MicroRNAs Provide Novel Insights into Their Regulation. J. Virol. 2011, 85, 996–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, A.; Makarewicz, O.; Egerer, R.; Karrasch, M.; Klink, A.; Sauerbrei, A.; Kentouche, K.; Pletz, M.W. EBV miRNA expression profiles in different infection stages: A prospective cohort study. PLoS ONE 2019, 14, e0212027. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Haar, J.; Bernhardt, K.; Linnstaedt, S.D.; Bannert, H.; Lips, H.; Cullen, B.R.; Delecluse, H.-J. The Members of an Epstein-Barr Virus MicroRNA Cluster Cooperate to Transform B Lymphocytes. J. Virol. 2011, 85, 9801–9810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Nie, K.; Redmond, D.; Liu, Y.; Elemento, O.; Knowles, D.M.; Tam, W. EBV-miR-BHRF1-2 targets PRDM1/Blimp1: Potential role in EBV lymphomagenesis. Leukemia 2016, 30, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31, 2207–2221. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Albanese, M.; Tagawa, T.; Buschle, A.; Hammerschmidt, W. Innate and Adaptive Antiviral Immunity. J. Virol. 2017, 91, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, K.; Haar, J.; Tsai, M.H.; Poirey, R.; Feederle, R.; Delecluse, H.J. A Viral microRNA Cluster Regulates the Expression of PTEN, p27 and of a bcl-2 Homolog. PLoS Pathog. 2016, 12, e1005405. [Google Scholar] [CrossRef]
- Skinner, C.M.; Ivanov, N.S.; Barr, S.A.; Chen, Y.; Skalsky, R.L. An Epstein-Barr Virus MicroRNA Blocks Interleukin-1 (IL-1) Signaling by Targeting IL-1 Receptor 1. J. Virol. 2017, 91, e00530-17. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Callegari, S.; Masucci, M.G. The Epstein-Barr virus miR-BHRF1-1 targets RNF4 during productive infection to promote the accumulation of SUMO conjugates and the release of infectious virus. PLoS Pathog. 2017, 13, e1006338. [Google Scholar] [CrossRef]
- Matsuda, S.; Koyasu, S. Mechanisms of action of cyclosporine. Immunopharmacology 2000, 47, 119–125. [Google Scholar] [CrossRef]
- Aalto, S.M.; Juvonen, E.; Tarkkanen, J.; Volin, L.; Ruutu, T.; Mattila, P.S.; Piiparinen, H.; Knuutila, S.; Hedman, K. Lymphoproliferative disease after allogeneic stem cell transplantation—Pre-Emptive diagnosis by quantification of Epstein-Barr virus DNA in serum. J. Clin. Virol. 2003, 28, 275–283. [Google Scholar] [CrossRef]
- Walker, I.; Panzarella, T.; Couban, S.; Couture, F.; Devins, G.; Elemary, M.; Gallagher, G.; Kerr, H.; Kuruvilla, J.; Lee, S.J.; et al. Pretreatment with anti-thymocyte globulin versus no anti-thymocyte globulin in patients with haematological malignancies undergoing haemopoietic cell transplantation from unrelated donors: A randomised, controlled, open-label, phase 3, multicentre trial. Lancet Oncol. 2016, 17, 164–173. [Google Scholar] [CrossRef]
- Elion, G.B. Mechanism of action and selectivity of acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Hanto, D.W.; Frizzera, G.; Gajl-Peczalska, K.J.; Sakamoto, K.; Purtilo, D.T.; Balfour, H.H.; Simmons, R.L.; Najarian, J.S. Epstein–Barr Virus-Induced B-Cell Lymphoma after Renal Transplantation. N. Engl. J. Med. 1982, 306, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Gershburg, E.; Pagano, J.S. Epstein-Barr virus infections: Prospects for treatment. J. Antimicrob. Chemother. 2005, 56, 277–281. [Google Scholar] [CrossRef]
- Gutiérrez, M.I.; Judde, J.G.; Magrath, I.T.; Bhatia, K.G. Switching viral latency to viral lysis: A novel therapeutic approach for Epstein-Barr virus-associated neoplasia. Cancer Res. 1996, 56, 969–972. [Google Scholar]
- Feng, W.; Hong, G.; Delecluse, H.-J.; Kenney, S.C. Lytic Induction Therapy for Epstein-Barr Virus-Positive B-Cell Lymphomas. J. Virol. 2004, 78, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Perrine, S.P.; Hermine, O.; Small, T.; Suarez, F.; O’Reilly, R.; Boulad, F.; Fingeroth, J.; Askin, M.; Levy, A.; Mentzer, S.J.; et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007, 109, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Heslop, H.E.; Ng, C.Y.C.; Li, C.; Smith, C.A.; Loftin, S.K.; Krance, R.; Brenner, M.K.; Rooney, C.M. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat. Med. 1996, 2, 551–555. [Google Scholar] [CrossRef]
- Gottschalk, S.; Rooney, C.M. Adoptive T-cell immunotherapy. Curr. Top. Microbiol. Immunol. 2015, 391, 427–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowska, J.; Stuehler, C.; Egli, A.; Battegay, M.; Rauser, G.; Bantug, G.R.; Brander, C.; Hess, C.; Khanna, N. T cells specific for different latent and lytic viral proteins efficiently control Epstein-Barr virus-transformed B cells. Cytotherapy 2015, 17, 1280–1291. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Immunomodulation and Immune Evasion | |||||
|---|---|---|---|---|---|
| EBV Lytic Gene | IE/E/L 1 | Lytic Function | Role in Oncogenesis | Oncogenic Mechanism of Action | References |
| BZLF1 | IE | Transactivator | Induction of pro-inflammatory cytokine expression and secretion (IL-8, IL-10, IL-13) | Binding and activating target gene promoters | [49,50,51] |
| BGLF5 | E | Alkaline exonuclease | Downregulation of MHCs | Host shut off; degradation of cellular mRNAs | [52] |
| BILF1 | E | gp64, vGPCR | Inhibition of MHC trafficking | [53] | |
| BLLF3 | E | dUTPase | Induction of pro-inflammatory cytokine expression and secretion (IL-1β, IL-6, IL-8, IL-10) | [54] | |
| BNLF2a | E | Inhibitor of TAP 2 | Inhibition of CD8 T cell recognition of infected cells | [55] | |
| BCRF1 | L | vIL-10 | Inhibition of NK cell-mediated elimination of infected cells; inhibition of CD4 T cells | [55] | |
| BDLF3 | L | gp150 | Downregulation of MHCs | Ubiquitination and degradation of MHCs | [56] |
| BZLF2 | L | gp42 | Inhibition of MHC II-mediated antigen presentation | [57] | |
| Angiogenesis and Invasion | |||||
| EBV Lytic Gene | IE/E/L | Lytic Function | Role in Oncogenesis | Oncogenic Mechanism of Action | References |
| BZLF1 | IE | Transactivator | Upregulation of MMP1, MMP3, MMP9 | Binding and activating target gene promoters | [58,59,60] |
| BRLF1 | IE | Transactivator | Upregulation of MMP9 | Binding and activating target gene promoters | [61] |
| Genomic Instability | |||||
| EBV Lytic Gene | IE/E/L | Lytic Function | Role in Oncogenesis | Oncogenic Mechanism of Action | References |
| BALF3 | E | Terminase | Induction of genomic aberration | Induction of DNA damage | [62] |
| BGLF4 | E | S/T protein kinase | Induction of genomic aberration | Induction of DNA damage pathways and premature chromosome condensation | [63,64] |
| BGLF5 | E | Alkaline exonuclease | Induction of genomic aberration | Induction of DNA damage | [65] |
| BALF4 | L | gp110 | Induction of genomic aberration | [66] | |
| BNRF1 | L | Major tegument protein | Induction of genomic aberration | [66] | |
| Cell Cycle Progression and Apoptosis | |||||
| EBV Lytic Gene | IE/E/L | Lytic Function | Role in Oncogenesis | Oncogenic Mechanism of Action | References |
| BALF1 | E | vBcl-2 | Pro-survival, anti-apoptotic | [67] | |
| BHRF1 | E | vBcl-2 | Pro-survival, anti-apoptotic | Inhibition of BIM, PUMA, BAK | [67,68,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. https://doi.org/10.3390/microorganisms8111824
Rosemarie Q, Sugden B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms. 2020; 8(11):1824. https://doi.org/10.3390/microorganisms8111824
Chicago/Turabian StyleRosemarie, Quincy, and Bill Sugden. 2020. "Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis" Microorganisms 8, no. 11: 1824. https://doi.org/10.3390/microorganisms8111824
APA StyleRosemarie, Q., & Sugden, B. (2020). Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms, 8(11), 1824. https://doi.org/10.3390/microorganisms8111824