Behind Taxonomic Variability: The Functional Redundancy in the Tick Microbiome




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Processing of Original Raw Sequences
2.2. Prediction of Functional Traits in Tick Microbiome
2.3. Differential Taxonomic Composition Analysis
2.4. Testing Functional Redundancy of Tick Microbiome
2.5. Co-Occurring Bacteria and Functional Similarity
3. Results
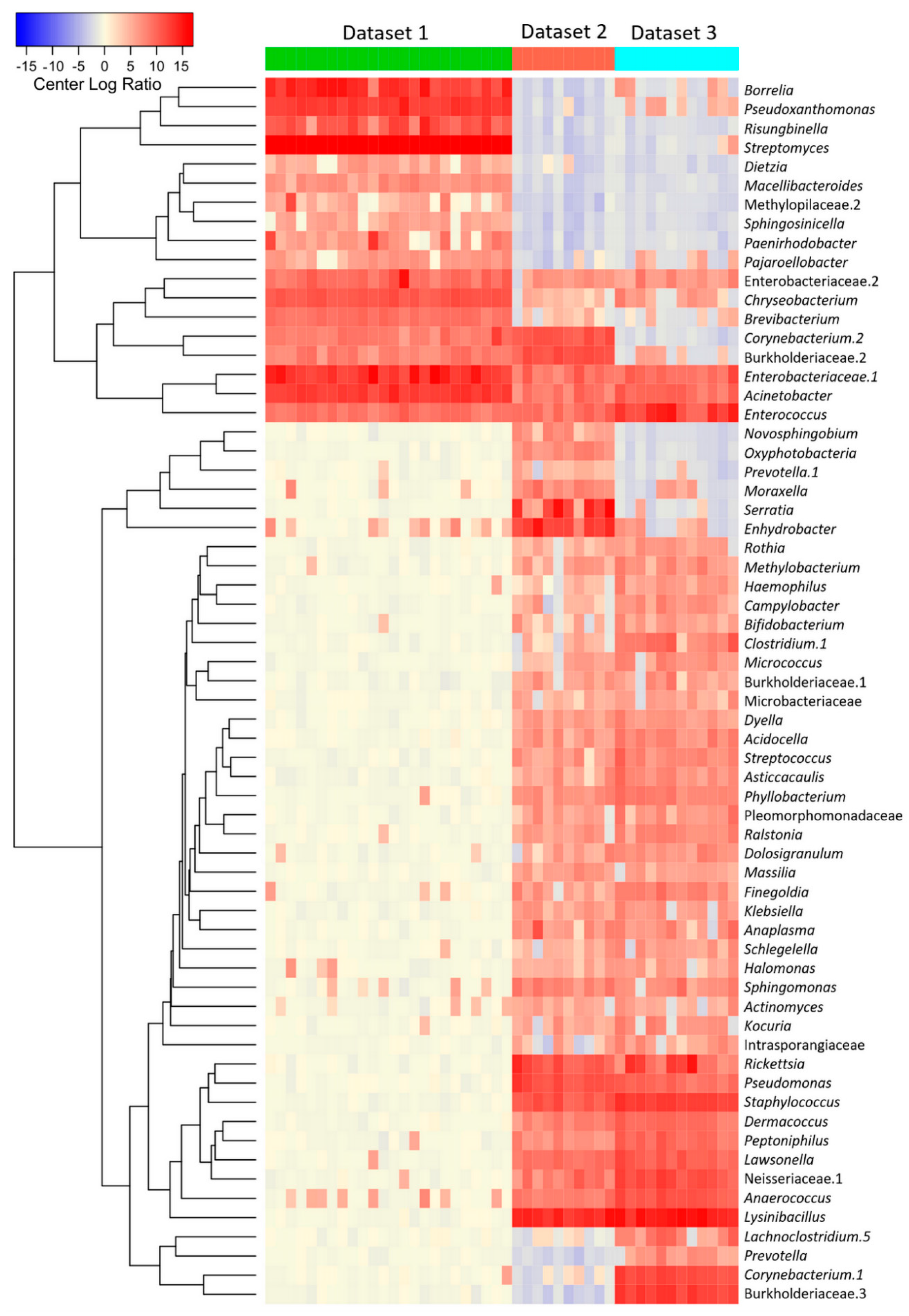
3.1. B. burgdorferi Infection and Developmental Stage Are Associated with Changes in Taxonomic Composition and Bacterial Abundance
3.2. Lower Functional Diversity in the Microbiome of Borrelia Infected Larvae Compared to Uninfected Nymphs
3.3. Taxa Diversity Contributes to Similar Functional Profiles in Tick Microbiome Regardless of Borrelia Infection and Developmental Stage
3.4. Functional Pathways Are Highly Redundant in the I. scapularis Microbiome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karim, S.; Budachetri, K.; Mukherjee, N.; Williams, J.; Kausar, A.; Hassan, M.J.; Adamson, S.; Dow, S.E.; Apanaskevich, D.; Arijo, A.; et al. A study of ticks and tick-borne livestock pathogens in Pakistan. PLoS Negl. Trop. Dis. 2017, 11, e0005681. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Abe, T.; Nijhof, A.M.; Yamamoto, S.; Jongejan, F.; Ikemura, T.; Sugimoto, C. A novel approach, based on BLSOMs (Batch Learning Self-Organizing Maps), to the microbiome analysis of ticks. ISME J. 2013, 7, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Sánchez, S.; Hernández-Jarguín, A.; Torina, A.; García Fernández de Mera, I.; Blanda, V.; Caracappa, S.; Gortazar, C.; de la Fuente, J. Characterization of the bacterial microbiota in wild-caught Ixodes ventalloi. Ticks Tick Borne Dis. 2019, 10, 336–343. [Google Scholar] [CrossRef]
- Budachetri, K.; Browning, R.E.; Adamson, S.W.; Dowd, S.E.; Chao, C.-C.; Ching, W.M.; Karim, S. An insight into the microbiome of the Amblyomma maculatum (Acari: Ixodidae). J. Med. Entomol. 2014, 51, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Budachetri, K.; Williams, J.; Mukherjee, N.; Sellers, M.; Moore, F.; Karim, S. The microbiome of neotropical ticks parasitizing on passerine migratory birds. Ticks Tick Borne Dis. 2017, 8, 170–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budachetri, K.; Gaillard, D.; Williams, J.; Mukherjee, N.; Karim, S. A snapshot of the microbiome of Amblyomma tuberculatum ticks infesting the gopher tortoise, an endangered species. Ticks Tick Borne Dis. 2016, 7, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Ross, B.D.; Hayes, B.; Radey, M.C.; Lee, X.; Josek, T.; Bjork, J.; Neitzel, D.; Paskewitz, S.; Chou, S.; Mougous, J.D. Ixodes scapularis does not harbor a stable midgut microbiome. ISME J. 2018, 12, 2596–2607. [Google Scholar] [CrossRef] [Green Version]
- Clow, K.M.; Weese, J.S.; Rousseau, J.; Jardine, C.M. Microbiota of field-collected Ixodes scapularis and Dermacentor variabilis from eastern and southern Ontario, Canada. Ticks Tick Borne Dis. 2018, 9, 235–244. [Google Scholar] [CrossRef]
- Yan, P.; Qiu, Z.; Zhang, T.; Li, Y.; Wang, W.; Li, M.; Yu, Z.; Liu, J. Microbial diversity in the tick Argas japonicus (Acari: Argasidae) with a focus on Rickettsia pathogens. Med. Vet. Entomol. 2019, 33, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Binetruy, F.; Dupraz, M.; Buysse, M.; Duron, O. Surface sterilization methods impact measures of internal microbial diversity in ticks. Parasit. Vectors 2019, 28, 268. [Google Scholar] [CrossRef] [Green Version]
- Lejal, E.; Estrada-Peña, A.; Marsot, M.; Cosson, J.-F.; Rué, O.; Mariadassou, M.; Midoux, C.; Vayssier-Taussat, M.; Pollet, T. Taxon appearance from extraction and amplification steps demonstrates the value of multiple controls in tick microbiota analysis. Front. Microbiol. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Cabezas-Cruz, A.; Pollet, T.; Vayssier-Taussat, M.; Cosson, J.-F. High-throughput sequencing and network analysis disentangle the microbial communities of ticks and hosts within and between ecosystems. Front. Cell. Infect. Microbiol. 2018, 8, 236. [Google Scholar] [CrossRef] [Green Version]
- Chicana, B.; Couper, L.I.; Kwan, J.Y.; Tahiraj, E.; Swei, A. Comparative microbiome profiles of sympatric tick species from the far-western United States. Insects 2019, 10, 353. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, C.A.; Reif, K.E.; Scoles, G.A.; Mason, K.L.; Mousel, M.; Noh, S.M.; Brayton, K.A. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J. 2016, 10, 1846–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; Deponte, K.; Fish, D.; Fikrig, E. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, N.A.; Sloan, D.B. The Hologenome concept: Helpful or hollow? PLoS Biol. 2015, 13, e1002311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.W.; Kohl, K.D.; Brucker, R.M.; van Opstal, E.J.; Bordenstein, S.R. Phylosymbiosis: Relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 2016, 14, e2000225. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Theis, K.R. Host biology in light of the microbiome: Ten principles of holobionts and hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Peña, A.; Cabezas-Cruz, A.; Obregón, D. Resistance of tick gut microbiome to anti-tick vaccines, pathogen infection and antimicrobial peptides. Pathogens 2020, 9, 309. [Google Scholar] [CrossRef]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Fikrig, E. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. Manipulation of FASTQ data with galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 1–5. [Google Scholar] [CrossRef]
- Bastian, M.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International Conference on Weblogs and Social Media, ICWSM 2009, San Jose, CA, USA, 17–20 May 2009; pp. 4–6. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disz, T.; Akhter, S.; Cuevas, D. Accessing the SEED Genome Databases via Web Services API: Tools for Programmers. BMC Bioinform. 2010, 11, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Fernandes, A.D. Displaying Variation in Large Datasets: A Visual Summary of Effect Sizes. J. Comp. Graph. Stat. 2016, 25, 971–979. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 19 April 2020).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R package version 2.0–7. 2013. Available online: http://CRAN.R-project.org/package=vegan (accessed on 19 November 2019).
- Louca, S.; Polz, M.F.; Mazel, F.; Albright, M.B.N.; Huber, J.A.; O’Connor, M.I.; Ackermann, M.; Hahn, A.S.; Srivastava, D.S.; Crowe, S.A.; et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2018, 2, 936–943. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 10. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Levy, R.; Borenstein, E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc. Natl. Acad. Sci. USA 2013, 110, 12804–12809. [Google Scholar] [CrossRef] [Green Version]
- Bouchon, D.; Zimmer, M.; Dittmer, J. The terrestrial isopod microbiome: An all-in-one toolbox for animal-microbe interactions of ecological relevance. Front. Microbiol. 2016, 7, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, C.; Steinberg, P.; Rusch, D.; Kjelleberg, S.; Thomas, T. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. USA 2011, 108, 14288–14293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The tick microbiome: Why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Duron, O.; Morel, O.; Noël, V.; Buysse, M.; Binetruy, F.; Lancelot, R.; Vial, L. Tick-bacteria mutualism depends on B vitamin synthesis pathways. Curr. Biol. 2018, 28, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.I.; Bateman, A.C.; Bik, H.M.; Meadow, J.F. Microbiota of the indoor environment: A meta-analysis. Microbiome 2015, 3, 49. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, I.; Mendel, J.; Šikutová, S.; Švec, P.; Masaříková, J.; Nováková, D.; Hubálek, Z. 16S rRNA gene-based identification of cultured bacterial flora from host-seeking Ixodes ricinus, Dermacentor reticulatus and Haemaphysalis concinna ticks, vectors of vertebrate pathogens. Folia Microbiol. 2009, 54, 419. [Google Scholar] [CrossRef]
- Egyed, L.; Makrai, L. Cultivable internal bacterial flora of ticks isolated in Hungary. Exp. Appl. Acarol. 2014, 63, 107–122. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada-Peña, A.; Cabezas-Cruz, A.; Obregón, D. Behind Taxonomic Variability: The Functional Redundancy in the Tick Microbiome. Microorganisms 2020, 8, 1829. https://doi.org/10.3390/microorganisms8111829
Estrada-Peña A, Cabezas-Cruz A, Obregón D. Behind Taxonomic Variability: The Functional Redundancy in the Tick Microbiome. Microorganisms. 2020; 8(11):1829. https://doi.org/10.3390/microorganisms8111829
Chicago/Turabian StyleEstrada-Peña, Agustín, Alejandro Cabezas-Cruz, and Dasiel Obregón. 2020. "Behind Taxonomic Variability: The Functional Redundancy in the Tick Microbiome" Microorganisms 8, no. 11: 1829. https://doi.org/10.3390/microorganisms8111829
APA StyleEstrada-Peña, A., Cabezas-Cruz, A., & Obregón, D. (2020). Behind Taxonomic Variability: The Functional Redundancy in the Tick Microbiome. Microorganisms, 8(11), 1829. https://doi.org/10.3390/microorganisms8111829