Type III Secretion Effectors with Arginine N-Glycosyltransferase Activity


Abstract
:1. Introduction
2. Glycosyltransferases
2.1. Classification
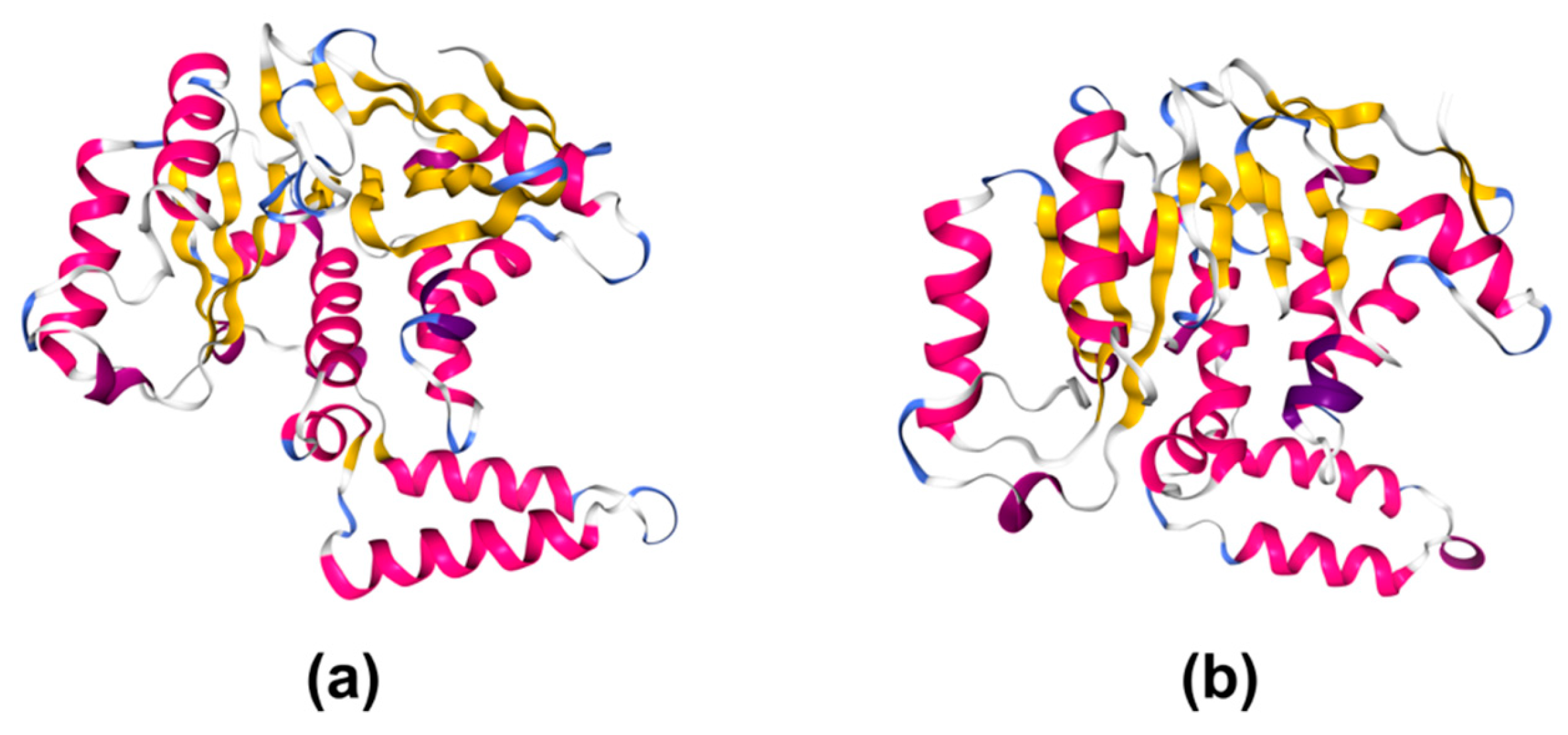
2.2. Structural Studies
3. Glycosylation in Bacterial Pathogenesis
3.1. Glycosylation of Adhesins
- A well-known example is the high-molecular-weight (HMW) system of Haemophilus influenza. HMW1/HMW2 are two adhesins that are glycosylated at the cytoplasm by GTs HMW1C and HMW2C, respectively [56,57]. Then, they are transported to the bacterial surface and mediate adherence to respiratory epithelial cells [57]. Glucose and galactose are added to specific asparagine residues in the eukaryotic-like Asn-X-Ser/Thr consensus [58].
- Pgl (from Protein glycosylation) system in Campylobacter jejuni is another example of a glycosylated adhesin that has been considered a prototype for the relevance of bacterial glycosylation in bacterial–host interaction [55]. It is an N-linked protein glycosylation system composed of several proteins, including various GTs that modify C. jejuni proteins leading to bacterial–host cell adhesion and invasion of host epithelium [59]. Disruption of the Pgl pathway results in reduction of human epithelial cells adhesion and decreases the ability to colonize mouse and chicken in vivo [59,60,61]. Modification catalyzed by the Pgl system consists of the addition of a heptasaccharide to asparagine residues within the eukaryotic-like Asn-X-Ser/Thr glycosylation consensus. The oligosaccharide is assembled on the cytoplasmic side of the inner membrane. Then, it is translocated by an ABC-transporter (PglK) [62]. Once in the periplasmic space, PglB, an oligo-saccharyltransferase, transfers the heptasaccharide to asparagine residues of target ligands [63,64].
- In addition to N-glycosylation-related mechanisms to improve bacterial adhesion, other systems based on O-glycosylation have been characterized. One example is the emerging bacterial autotransporter heptosyltransferase (BAHT) family found in several Gram-negative bacteria [65]. Autotransporters which belong to the T5SS family have a modular composition with an N-terminal passenger domain and a C-terminal β-barrel domain that drives the passenger across the outer membrane. The passenger domain may be heptosylated by a BAHT member on numerous serine residues. This modification occurs before translocation of the protein across the inner membrane and is required for bacterial adhesion to the host. Two well-characterized BAHT member are AAH (autotransporter adhesin heptosyltransferase), from diffusely adhering E. coli isolate 2787, and TibC from enterotoxigenic E. coli strain H10407 [65,66]. In addition to pathogenic E. coli, BAHT members have been identified in other bacteria, including Citrobacter, Shigella, Salmonella, and Burkholderia [65]. Further studies are necessary to fully characterize these interesting bacterial enzymes.
- Post-translational modification of bacterial proteins to enhance bacterial adhesion to the host is not restricted to Gram-negative bacteria. A well-known example of modified bacterial proteins in Gram-positive bacteria are the O-glycosylated serine-rich repeat proteins (SRRPs) [67]. These large glycoproteins, unique in Gram-positive bacteria, are crucial for biofilm formation and, more importantly, host cell adhesion [68]. The best-characterized SRRP is Fap1 from Streptococcus parasanguinis. Glycosylation of Fap1 relies on the activity of at least six different GTs, although the specific function and glycan transferred remains unclear [69]. This family of glycoproteins is highly conserved among streptococcal and staphylococcal species [67], thus, characterization of SRRPs GTs and understanding of their biological role are key to identify new therapeutic drug targets in order to tackle relevant Gram-positive pathogens.
3.2. Glycosylation of Flagellins
- In Aeromonas caviae Sch3N, the polar flagellins, FlaA and FlaB, are glycosylated with pseudoaminic acid glycans and this glycosylation is required for flagellar assembly. Genes involved in glycosylation are mapped in the O-antigen biosynthetic cluster [86,87]. In Aeromonas hydrophila AH-3, lateral flagellin is modified with a derivative of pseudaminic acid, whereas polar flagella are glycosylated with a heterogeneous glycan [88].
- C. jejuni FlaA is glycosylated at up to 19 sites that contribute to the flagellum serospecificity. FlaB is also glycosylated, but the number of sites remains to be established. The predominant O-glycans are derivatives of pseudoaminic acid and legionaminic acid. The genes encoding GTs involved in flagellin glycosylation are located adjacent to the flagellin structural genes, flaA and flaB [85,89,90].
- In Helicobacter pylori, both the FlaA and FlaB structural proteins are glycosylated with pseudomainic acid at seven and ten sites, respectively. Biosynthesis of pseudaminic acid has been extensively studied in H. pylori and C. jejuni and involves six consecutive enzymatic steps before being transferred onto flagellins via O-linked glycosylation [91,92].
- The GT transferring pseudaminic acid onto flagellin has been studied in Magnetospirillum magneticum AMB-1 [93]. This GT, known as Maf, displays significant sequence identity with proteins from other Gram-negative bacteria, including Maf1 from A. caviae and PseE from C. jejuni, that have been shown to transfer pseudaminic acid onto flagellins in these organisms [94,95]. The structure of M. magneticum Maf displays a modified GT-A topology as found in CAZy families GT29 and GT42. In fact, the closest structural homologue is C. jejuni sialyltransferase Cst-II (PDB 1RO7), belonging to family GT42 [93].
- Glycosyltation of Clostridium difficile flagellin is essential for motility. Two types of modification have been defined as type A and B. Type A modification involves an O-linked N-acetylglucosamine (GlcNAc) linked to a methylated threonine via a phosphodiester bond. Three GTs are involved in type B modification: GT1 and GT2 are responsible for the sequential addition of a GlcNAc and two rhamnoses, respectively, and GT3 is associated with the incorporation of a sulfonated peptidyl-amido sugar moiety [74].
3.3. Modification of Host Cell Targets by Bacterial GTs
- Large clostridial glycosylating toxins: pathogenesis of Clostridium depends on the activity of two similar toxins, TcdA and TcdB, that exhibit GT activity in their N-terminal domains [96,97]. Both toxins perform O-glycosylation of small GTPases of the Rho family, inhibiting the regulatory functions of these proteins [98]. This modification occurs on the side chain hydroxyl group of a threonine residue, which is specific for each targeted Rho GTPase [99]. Another example of this family of toxins recently identified is PaTox from entomopathogenic Photorhabdus asymbiotica [100].
- Legionella pneumophila Lgt family: L. pneumophila is the causative agent of legionellosis, a severe lung disease. This facultative intracellular pathogen uses a T4SS to inject more than 300 effectors into the host cell [101]. Among them, L. pneumophila secretes several GTs (Lgt1-3) that perform specific glycosylation of a serine residue (Ser53) in the eukaryotic elongation factor 1A, a large GTPase and a component of the elongation complex in protein synthesis. This modification results in inhibition of protein synthesis and subsequently causes the death of infected cells [102,103]. Interestingly, Lgt1 share high structural homology with the clostridial toxin TcdB. Both GTs are members of the clan III of retaining enzymes with the GT-A fold [103,104,105].
- NleB/SseK family: we refer to this family of T3SS effectors in the next section.
4. Glycosyltransferases of the NleB Family
4.1. Members of the Family
4.2. Expression, Translocation, and Subcellular Localization
4.3. Enzymatic Activity and Structural Studies
4.4. Role in Virulence
4.5. Targets of NleB/SseK Effectors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, S.; Grin, I.; Malmsheimer, S.; Singh, N.; Torres-Vargas, C.E.; Westerhausen, S. Bacterial type III secretion systems: A complex device for the delivery of bacterial effector proteins into eukaryotic host cells. FEMS Microbiol. Lett. 2018, 365, fny201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán, J.E.; Lara-Tejero, M.; Marlovits, T.C.; Wagner, S. Bacterial type III secretion systems: Specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 2014, 68, 415–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán, J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe 2009, 5, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Guttman, D.S. Evolution of prokaryotic and eukaryotic virulence effectors. Curr. Opin. Plant Biol. 2008, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.E.; Hartland, E.L. Post-translational mechanisms of host subversion by bacterial effectors. Trends Mol. Med. 2017, 23, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Brenner, F.W.; Villar, R.G.; Angulo, F.J.; Tauxe, R.; Swaminathan, B. Salmonella nomenclature. J. Clin. Microbiol. 2000, 38, 2465–2467. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Soncini, F.C.; Solomon, F.; Groisman, E.A. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7800–7804. [Google Scholar] [CrossRef] [Green Version]
- Shea, J.E.; Hensel, M.; Gleeson, C.; Holden, D.W. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 1996, 93, 2593–2597. [Google Scholar] [CrossRef] [Green Version]
- Galán, J.E.; Curtiss, R. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. USA 1989, 86, 6383–6387. [Google Scholar] [CrossRef] [Green Version]
- Fookes, M.; Schroeder, G.N.; Langridge, G.C.; Blondel, C.J.; Mammina, C.; Connor, T.R.; Seth-Smith, H.; Vernikos, G.S.; Robinson, K.S.; Sanders, M.; et al. Salmonella bongori provides insights into the evolution of the salmonellae. PLoS Pathog. 2011, 7, e1002191. [Google Scholar] [CrossRef] [Green Version]
- Lou, L.; Zhang, P.; Piao, R.; Wang, Y. Salmonella pathogenicity island 1 (SPI-1) and its complex regulatory network. Front. Cell. Infect. Microbiol. 2019, 9, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuli, A.; Sharma, M. How to do business with lysosomes: Salmonella leads the way. Curr. Opin. Microbiol. 2019, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jennings, E.; Thurston, T.L.M.; Holden, D.W. Salmonella SPI-2 type III secretion system effectors: Molecular mechanisms and physiological consequences. Cell Host Microbe 2017, 22, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Habyarimana, F.; Sabag-Daigle, A.; Ahmer, B.M.M. The SdiA-regulated gene srgE encodes a type III secreted effector. J. Bacteriol. 2014, 196, 2301–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Kamanova, J.; Lara-Tejero, M.; Galán, J.E. A family of Salmonella type III secretion effector proteins selectively targets the NF-κB signaling pathway to preserve host homeostasis. PLoS Pathog. 2016, 12, e1005484. [Google Scholar] [CrossRef]
- Jaslow, S.L.; Gibbs, K.D.; Fricke, W.F.; Wang, L.; Pittman, K.J.; Mammel, M.K.; Thaden, J.T.; Fowler, V.G.; Hammer, G.E.; Elfenbein, J.R.; et al. Salmonella activation of STAT3 signaling by SarA effector promotes intracellular replication and production of IL-10. Cell Rep. 2018, 23, 3525–3536. [Google Scholar] [CrossRef]
- Ramos-Morales, F. Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. 2012, 2012, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Frankel, G.; Phillips, A.D.; Rosenshine, I.; Dougan, G.; Kaper, J.B.; Knutton, S. Enteropathogenic and enterohaemorrhagic Escherichia coli: More subversive elements. Mol. Microbiol. 1998, 30, 911–921. [Google Scholar] [CrossRef]
- Barthold, S.W. The microbiology of transmissible murine colonic hyperplasia. Lab. Anim. Sci. 1980, 30, 167–173. [Google Scholar]
- Deng, W.; Li, Y.; Vallance, B.A.; Finlay, B.B. Locus of enterocyte effacement from Citrobacter rodentium: Sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Infect. Immun. 2001, 69, 6323–6335. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, K.G.; Girón, J.A.; Jerse, A.E.; Mcdaniel, T.K.; Donnenberg, M.S.; Kaper, J.B. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. USA 1995, 92, 7996–8000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaytán, M.O.; Martínez-Santos, V.I.; Soto, E.; González-Pedrajo, B. Type three secretion system in attaching and effacing pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Guo, Z.; Duan, Q.; Hardwidge, P.R.; Zhu, G. Escherichia coli type III secretion system 2: A new kind of T3SS? Vet. Res. 2014, 45, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete genome sequence of enterohemorrhagic Eschelichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Shulman, A.; Yair, Y.; Biran, D.; Sura, T.; Otto, A.; Gophna, U.; Becher, D.; Hecker, M.; Ron, E.Z. The Escherichia coli type III secretion system 2 has a global effect on cell surface. MBio 2018, 9, e01070-18. [Google Scholar] [CrossRef] [Green Version]
- Ooka, T.; Ogura, Y.; Katsura, K.; Seto, K.; Kobayashi, H.; Kawano, K.; Tokuoka, E.; Furukawa, M.; Harada, S.; Yoshino, S.; et al. Defining the genome features of Escherichia albertii, an emerging enteropathogen closely related to Escherichia coli. Genome Biol. Evol. 2015, 7, 3170–3179. [Google Scholar]
- Ideses, D.; Gophna, U.; Paitan, Y.; Chaudhuri, R.R.; Pallen, M.J.; Ron, E.Z. A degenerate type III secretion system from septicemic Escherichia coli contributes to pathogenesis. J. Bacteriol. 2005, 187, 8164–8171. [Google Scholar] [CrossRef] [Green Version]
- Hurley, D.; McCusker, M.P.; Fanning, S.; Martins, M. Salmonella-host interactions—Modulation of the host innate immune system. Front. Immunol. 2014, 5, 481. [Google Scholar] [CrossRef] [Green Version]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [Green Version]
- Mestrom, L.; Przypis, M.; Kowalczykiewicz, D.; Pollender, A.; Kumpf, A.; Marsden, S.R.; Bento, I.; Jarzębski, A.B.; Szymańska, K.; Chruściel, A.; et al. Leloir glycosyltransferases in applied biocatalysis: A multidisciplinary approach. Int. J. Mol. Sci. 2019, 20, 5263. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Sinnott, M.L. Catalytic mechanism of enzymic glycosyl transfer. Chem. Rev. 1990, 90, 1171–1202. [Google Scholar] [CrossRef]
- Liang, D.M.; Liu, J.H.; Wu, H.; Wang, B. Bin; Zhu, H.J.; Qiao, J.J. Glycosyltransferases: Mechanisms and applications in natural product development. Chem. Soc. Rev. 2015, 44, 8350–8374. [Google Scholar] [CrossRef]
- Davies, G.J. Sweet secrets of synthesis. Nat. Struct. Biol. 2001, 8, 98–100. [Google Scholar] [CrossRef]
- Vocadlo, D.J.; Davies, G.J.; Laine, R.; Withers, S.G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar] [CrossRef]
- Soya, N.; Fang, Y.; Palcic, M.M.; Klassen, J.S. Trapping and characterization of covalent intermediates of mutant retaining glycosyltransferases. Glycobiology 2011, 21, 547–552. [Google Scholar] [CrossRef]
- Lira-Navarrete, E.; Iglesias-Fernández, J.; Zandberg, W.F.; Compañón, I.; Kong, Y.; Corzana, F.; Pinto, B.M.; Clausen, H.; Peregrina, J.M.; Vocadlo, D.J.; et al. Substrate-guided front-face reaction revealed by combined structural snapshots and metadynamics for the polypeptide N-acetylgalactosaminyltransferase 2. Angew. Chem. Int. Ed. Engl. 2014, 53, 8206–8210. [Google Scholar] [CrossRef]
- Ardèvol, A.; Rovira, C. The molecular mechanism of enzymatic glycosyl transfer with retention of configuration: Evidence for a short-lived oxocarbenium-like species. Angew. Chem. Int. Ed. 2011, 50, 10897–10901. [Google Scholar] [CrossRef]
- Schuman, B.; Evans, S.V.; Fyles, T.M. Geometric attributes of retaining glycosyltransferase enzymes favor an orthogonal mechanism. PLoS ONE 2013, 8, e71077. [Google Scholar] [CrossRef] [Green Version]
- Dodson, E.; Harding, M.M.; Hodgkin, D.C.; Rossmann, M.G. The crystal structure of insulin: III. Evidence for a 2-fold axis in rhombohedral zinc insulin. J. Mol. Biol. 1966, 16, 227–241. [Google Scholar] [CrossRef]
- Schuman, B.; Alfaro, J.A.; Evans, S.V. Glycosyltransferase Structure and Function. In Bioactive Conformation I; Springer: Berlin/Heidelberg, Germany, 2006; pp. 217–257. [Google Scholar]
- Kikuchi, N.; Kwon, Y.D.; Gotoh, M.; Narimatsu, H. Comparison of glycosyltransferase families using the profile hidden Markov model. Biochem. Biophys. Res. Commun. 2003, 310, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mushegian, A. Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci. 2003, 12, 1418–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosén, M.L.; Edman, M.; Sjöström, M.; Wieslander, A. Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence analysis. J. Biol. Chem. 2004, 279, 38683–38692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizak, C.; Gerber, S.; Numao, S.; Aebi, M.; Locher, K.P. X-ray structure of a bacterial oligosaccharyltransferase. Nature 2011, 474, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, F.; Yang, T.; Ding, L.; Zhou, M.; Li, J.; Haslam, S.M.; Dell, A.; Erlandsen, H.; Wu, H. The highly conserved domain of unknown function 1792 has a distinct glycosyltransferase fold. Nat. Commun. 2014, 5, 4339. [Google Scholar] [CrossRef] [Green Version]
- Lovering, A.L.; De Castro, L.H.; Lim, D.; Strynadka, N.C.J. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science (80-) 2007, 315, 1402–1405. [Google Scholar] [CrossRef]
- Yuan, Y.; Barrett, D.; Zhang, Y.; Kahne, D.; Sliz, P.; Walker, S. Crystal structure of a peptidoglycan glycosyltransferase suggests a model for processive glycan chain synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 5348–5353. [Google Scholar] [CrossRef] [Green Version]
- Kattke, M.D.; Gosschalk, J.E.; Martinez, O.E.; Kumar, G.; Gale, R.T.; Cascio, D.; Sawaya, M.R.; Philips, M.; Brown, E.D.; Clubb, R.T. Structure and mechanism of TagA, a novel membrane-associated glycosyltransferase that produces wall teichoic acids in pathogenic bacteria. PLoS Pathog. 2019, 15, e1007723. [Google Scholar] [CrossRef] [Green Version]
- Charnock, S.J.; Davies, G.J. Structure of the nucleotide-diphospho-sugar transferase, SpsA from Bacillus subtilis, in native and nucleotide-complexed forms. Biochemistry 1999, 38, 6380–6385. [Google Scholar] [CrossRef]
- Mulichak, A.M.; Losey, H.C.; Walsh, C.T.; Garavito, R.M. Structure of the UDP-glucosyltransferase GtfB that modifies the heptapeptide aglycone in the biosynthesis of vancomycin group antibiotics. Structure 2001, 9, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Li, S.; Shao, F. Sweet talk: Protein glycosylation in bacterial interaction with the host. Trends Microbiol. 2015, 23, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothaft, H.; Szymanski, C.M. Protein glycosylation in bacteria: Sweeter than ever. Nat. Rev. Microbiol. 2010, 8, 765–778. [Google Scholar] [CrossRef]
- Grass, S.; Buscher, A.Z.; Swords, W.E.; Apicella, M.A.; Barenkamp, S.J.; Ozchlewski, N.; St Geme, J.W. The Haemophilus influenzae HMW1 adhesin is glycosylated in a process that requires HMW1C and phosphoglucomutase, an enzyme involved in lipooligosaccharide biosynthesis. Mol. Microbiol. 2003, 48, 737–751. [Google Scholar] [CrossRef] [Green Version]
- St Geme, J.W.; Grass, S. Secretion of the Haemophilus influenzae HMW1 and HMW2 adhesins involves a periplasmic intermediate and requires the HMWB and HMWC proteins. Mol. Microbiol. 1998, 27, 617–630. [Google Scholar] [CrossRef]
- Gross, J.; Grass, S.; Davis, A.E.; Gilmore-Erdmann, P.; Townsend, R.R.; St. Geme, J.W. The Haemophilus influenzae HMW1 adhesin is a glycoprotein with an unusual N-linked carbohydrate modification. J. Biol. Chem. 2008, 283, 26010–26015. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, C.M.; Burr, D.H.; Guerry, P. Campylobacter protein glycosylation affects host cell interactions. Infect. Immun. 2002, 70, 2242–2244. [Google Scholar] [CrossRef] [Green Version]
- Karlyshev, A.V.; Everest, P.; Linton, D.; Cawthraw, S.; Newell, D.G.; Wren, B.W. The Campylobacter jejuni general glycosylation system is important for attachment to human epithelial cells and in the colonization of chicks. Microbiology 2004, 150, 1957–1964. [Google Scholar] [CrossRef] [Green Version]
- Hendrixson, D.R.; DiRita, V.J. Identification of Campylobacter jejuni genes involved in commensal colonization of the chick gastrointestinal tract. Mol. Microbiol. 2004, 52, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Perez, C.; Gerber, S.; Boilevin, J.; Bucher, M.; Darbre, T.; Aebi, M.; Reymond, J.-L.; Locher, K.P. Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 2015, 524, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Wacker, M.; Linton, D.; Hitchen, P.G.; Nita-Lazar, M.; Haslam, S.M.; North, S.J.; Panico, M.; Morris, H.R.; Dell, A.; Wren, B.W.; et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 2002, 298, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, H.; Maryam, A.; Shahid, R.; Siddiqi, A.R. Oligosaccharyltransferase PglB of Campylobacter jejuni is a glycoprotein. World J. Microbiol. Biotechnol. 2020, 36, 9. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yao, Q.; Xu, Y.; Li, L.; Li, S.; Liu, Y.; Gao, W.; Niu, M.; Sharon, M.; Ben-Nissan, G.; et al. An iron-containing dodecameric heptosyltransferase family modifies bacterial autotransporters in pathogenesis. Cell Host Microbe 2014, 16, 351–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benz, I.; Schmidt, M.A. Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol. Microbiol. 2001, 40, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Wu, H. Glycosylation and biogenesis of a family of serine-rich bacterial adhesins. Microbiology 2009, 155, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, A.; Sanchez, C.J.; Orihuela, C.J. A role for glycosylated serine-rich repeat proteins in gram-positive bacterial pathogenesis. Mol. Oral Microbiol. 2012, 27, 257–269. [Google Scholar] [CrossRef]
- Zhu, F.; Zhang, H.; Yang, T.; Haslam, S.M.; Dell, A.; Wu, H. Engineering and dissecting the glycosylation pathway of a streptococcal serine-rich repeat adhesin. J. Biol. Chem. 2016, 291, 27354–27363. [Google Scholar] [CrossRef]
- Logan, S.M. Flagellar glycosylation—A new component of the motility repertoire? Microbiology 2006, 152, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhou, M.; Zhu, L.; Zhu, G. Flagella and bacterial pathogenicity. J. Basic Microbiol. 2013, 53, 1–8. [Google Scholar] [CrossRef]
- Merino, S.; Tomás, J.M. Gram-negative flagella glycosylation. Int. J. Mol. Sci. 2014, 15, 2840–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvaux, N.A.; Thoden, J.B.; Holden, H.M. Molecular architectures of Pen and Pal: Key enzymes required for CMP-pseudaminic acid biosynthesis in Bacillus thuringiensis. Protein Sci. 2018, 27, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Valiente, E.; Bouché, L.; Hitchen, P.; Faulds-Pain, A.; Songane, M.; Dawson, L.F.; Donahue, E.; Stabler, R.A.; Panico, M.; Morris, H.R.; et al. Role of glycosyltransferases modifying type B flagellin of emerging hypervirulent Clostridium difficile lineages and their impact on motility and biofilm formation. J. Biol. Chem. 2016, 291, 25450–25461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouché, L.; Panico, M.; Hitchen, P.; Binet, D.; Sastre, F.; Faulds-Pain, A.; Valiente, E.; Vinogradov, E.; Aubry, A.; Fulton, K.; et al. The type B flagellin of hypervirulent Clostridium difficile is modified with novel sulfonated peptidylamido-glycans. J. Biol. Chem. 2016, 291, 25439–25449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maayer, P.; Cowan, D.A. Comparative genomic analysis of the flagellin glycosylation island of the Gram-positive thermophile Geobacillus. BMC Genom. 2016, 17, 913. [Google Scholar] [CrossRef] [Green Version]
- Janesch, B.; Schirmeister, F.; Maresch, D.; Altmann, F.; Messner, P.; Kolarich, D.; Schäffer, C. Flagellin glycosylation in Paenibacillus alvei CCM 2051T. Glycobiology 2016, 26, 74–87. [Google Scholar]
- Twine, S.M.; Reid, C.W.; Aubry, A.; McMullin, D.R.; Fulton, K.M.; Austin, J.; Logan, S.M. Motility and flagellar glycosylation in Clostridium difficile. J. Bacteriol. 2009, 191, 7050–7062. [Google Scholar] [CrossRef] [Green Version]
- Twine, S.M.; Paul, C.J.; Vinogradov, E.; McNally, D.J.; Brisson, J.R.; Mullen, J.A.; McMullin, D.R.; Jarrell, H.C.; Austin, J.W.; Kelly, J.F.; et al. Flagellar glycosylation in Clostridium botulinum. FEBS J. 2008, 275, 4428–4444. [Google Scholar] [CrossRef]
- Schirm, M.; Kalmokoff, M.; Aubry, A.; Thibault, P.; Sandoz, M.; Logan, S.M. Flagellin from Listeria monocytogenes is glycosylated with β-O-linked N-acetylglucosamine. J. Bacteriol. 2004, 186, 6721–6727. [Google Scholar] [CrossRef] [Green Version]
- Haya, S.; Tokumaru, Y.; Abe, N.; Kaneko, J.; Aizawa, S.I. Characterization of lateral flagella of Selenomonas ruminantium. Appl. Environ. Microbiol. 2011, 77, 2799–2802. [Google Scholar] [CrossRef] [Green Version]
- Merino, S.; Aquilini, E.; Fulton, K.M.; Twine, S.M.; Tomás, J.M. The polar and lateral flagella from Plesiomonas shigelloides are glycosylated with legionaminic acid. Front. Microbiol. 2015, 6, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.E.; Twine, S.M.; Fulton, K.M.; Titball, R.W.; Essex-Lopresti, A.E.; Atkins, T.P.; Prior, J.L. Flagellar glycosylation in Burkholderia pseudomallei and Burkholderia thailandensis. J. Bacteriol. 2011, 193, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Rath, C.B.; Schirmeister, F.; Figl, R.; Seeberger, P.H.; Schäffer, C.; Kolarich, D. Flagellin glycoproteomics of the periodontitis associated pathogen Selenomonas sputigena reveals previously not described O-glycans and rhamnose fragment rearrangement occurring on the glycopeptides. Mol. Cell. Proteom. 2018, 17, 721–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, P.; Logan, S.M.; Kelly, J.F.; Brisson, J.R.; Ewing, C.P.; Trust, T.J.; Guerry, P. Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin. J. Biol. Chem. 2001, 276, 34862–34870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.L.; Day-Williams, M.J.; Tomas, J.M.; Stafford, G.P.; Shaw, J.G. Identification of a putative glycosyltransferase responsible for the transfer of pseudaminic acid onto the polar flagellin of Aeromonas caviae Sch3N. Microbiologyopen 2012, 1, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.A.; Gryllos, I.; Tomás, J.M.; Shaw, J.G. Motility and the polar flagellum are required for Aeromonas caviae adherence to HEp-2 cells. Infect. Immun. 2001, 69, 4257–4267. [Google Scholar] [CrossRef] [Green Version]
- Wilhelms, M.; Fulton, K.M.; Twine, S.M.; Tomás, J.M.; Merino, S. Differential glycosylation of polar and lateral flagellins in Aeromonas hydrophila AH-3. J. Biol. Chem. 2012, 287, 27851–27862. [Google Scholar] [CrossRef] [Green Version]
- Dorrell, N.; Mangan, J.A.; Laing, K.G.; Hinds, J.; Linton, D.; Al-Ghusein, H.; Barrell, B.G.; Parkhill, J.; Stoker, N.G.; Karlyshev, A.V.; et al. Whole genome comparison of Campylobacter jejuni human isolates using a low-cost microarray reveals extensive genetic diversity. Genome Res. 2001, 11, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Alm, R.A.; Guerry, P.; Power, M.E.; Trust, T.J. Variation in antigenicity and molecular weight of Campylobacter coli VC167 flagellin in different genetic backgrounds. J. Bacteriol. 1992, 174, 4230–4238. [Google Scholar] [CrossRef] [Green Version]
- Schoenhofen, I.C.; McNally, D.J.; Brisson, J.-R.; Logan, S.M. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: Synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology 2006, 16, 8C–14C. [Google Scholar] [CrossRef]
- Schirm, M.; Soo, E.C.; Aubry, A.J.; Austin, J.; Thibault, P.; Logan, S.M. Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol. Microbiol. 2003, 48, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Sulzenbacher, G.; Roig-Zamboni, V.; Lebrun, R.; Guérardel, Y.; Murat, D.; Mansuelle, P.; Yamakawa, N.; Qian, X.X.; Vincentelli, R.; Bourne, Y.; et al. Glycosylate and move! The glycosyltransferase Maf is involved in bacterial flagella formation. Environ. Microbiol. 2018, 20, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, D.J.; Hui, J.P.M.; Aubry, A.J.; Mui, K.K.K.; Guerry, P.; Brisson, J.R.; Logan, S.M.; Soo, E.C. Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81–176 using a focused metabolomics approach. J. Biol. Chem. 2006, 281, 18489–18498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.L.; Lowry, R.C.; Couto, N.A.S.; Wright, P.C.; Stafford, G.P.; Shaw, J.G. Maf-dependent bacterial flagellin glycosylation occurs before chaperone binding and flagellar T3SS export. Mol. Microbiol. 2014, 92, 258–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef] [Green Version]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar] [CrossRef]
- Genth, H.; Aktories, K.; Just, I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J. Biol. Chem. 1999, 274, 29050–29056. [Google Scholar] [CrossRef] [Green Version]
- Jank, T.; Bogdanović, X.; Wirth, C.; Haaf, E.; Spoerner, M.; Böhmer, K.E.; Steinemann, M.; Orth, J.H.C.; Kalbitzer, H.R.; Warscheid, B.; et al. A bacterial toxin catalyzing tyrosine glycosylation of Rho and deamidation of Gq and Gi proteins. Nat. Struct. Mol. Biol. 2013, 20, 1273–1280. [Google Scholar] [CrossRef]
- Qiu, J.; Luo, Z.-Q. Legionella and Coxiella effectors: Strength in diversity and activity. Nat. Rev. Microbiol. 2017, 15, 591–605. [Google Scholar] [CrossRef]
- Belyi, Y.; Tabakova, I.; Stahl, M.; Aktories, K. Lgt: A family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 2008, 190, 3026–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, W.; Du, J.; Stahl, M.; Tzivelekidis, T.; Belyi, Y.; Gerhardt, S.; Aktories, K.; Einsle, O. Structural basis of the action of glucosyltransferase Lgt1 from Legionella pneumophila. J. Mol. Biol. 2010, 396, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Jank, T.; Aktories, K.; Schulz, G.E. Structural basis for the function of Clostridium difficile toxin B. J. Mol. Biol. 2005, 351, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Stahl, M.; Sovkova, I.; Kaden, P.; Luy, B.; Aktories, K. Region of elongation factor 1A1 involved in substrate recognition by Legionella pneumophila glucosyltransferase Lgt1. Identification of Lgt1 as a retaining glucosyltransferase. J. Biol. Chem. 2009, 284, 20167–20174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Puente, J.L.; Gruenheid, S.; Li, Y.; Vallance, B.A.; Vázquez, A.; Barba, J.; Ibarra, J.A.; O’Donnell, P.; Metalnikov, P.; et al. Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. USA 2004, 101, 3597–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, N.K.; Bulgin, R.; Crepin, V.F.; Cerdeño-Tárraga, A.M.; Schroeder, G.N.; Quail, M.A.; Lennard, N.; Corton, C.; Barron, A.; Clark, L.; et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 2010, 192, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, N.T.; Plunkett, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, A.; Thomson, N.R.; Ogura, Y.; Saunders, D.; Ooka, T.; Henderson, I.R.; Harris, D.; Asadulghani, M.; Kurokawa, K.; Dean, P.; et al. Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 2009, 91, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.; Hart, E.; Mundy, R.; Marchès, O.; Wiles, S.; Badea, L.; Luck, S.; Tauschek, M.; Frankel, G.; Robins-Browne, R.M.; et al. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect. Immun. 2006, 74, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Coombes, B.K.; Bishop, J.L.; Wickham, M.E.; Lowden, M.J.; Gal-Mor, O.; Goode, D.L.; Boyle, E.C.; Sanderson, K.L.; Finlay, B.B.; et al. Salmonella phage ST64B encodes a member of the SseK/NleB effector family. PLoS ONE 2011, 6, e17824. [Google Scholar] [CrossRef] [Green Version]
- Kujat Choy, S.L.; Boyle, E.C.; Gal-Mor, O.; Goode, D.L.; Valdez, Y.; Vallance, B.A.; Finlay, B.B. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar Typhimurium. Infect. Immun. 2004, 72, 5115–5125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvik, T.; Smillie, C.; Groisman, E.A.; Ochman, H. Short-term signatures of evolutionary change in the Salmonella enterica serovar Typhimurium 14028 genome. J. Bacteriol. 2010, 192, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswarappa, S.M.; Janice, J.; Balasundaram, S.V.; Chakravortty, D. Non-neutral evolution in non-LEE-encoded type III effectors of attaching and effacing Escherichia coli. Microbes Infect. 2013, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Egan, M.; Critelli, B.; Kouse, A.; Kalman, D.; Upreti, C. The evasive enemy: Insights into the virulence and epidemiology of the emerging attaching and effacing pathogen Escherichia albertii. Infect. Immun. 2019, 87, e00254-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Feng, J.; Pu, J.; Xu, X.; Lu, S.; Yang, J.; Wang, Y.; Jin, D.; Du, X.; Meng, X.; et al. Genomic and molecular characterisation of Escherichia marmotae from wild rodents in Qinghai-Tibet plateau as a potential pathogen. Sci. Rep. 2019, 9, 10619. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.V.; Muthappa, D.M.; Hurley, D.; Donoghue, O.; McCabe, E.; Anes, J.; Schaffer, K.; Murphy, B.P.; Buckley, J.F.; Fanning, S. Yersinia hibernica sp. nov., isolated from pig-production environments. Int. J. Syst. Evol. Microbiol. 2019, 69, 2023–2027. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.T.; Sperandio, V. Inter-kingdom signalling: Communication between bacteria and their hosts. Nat. Rev. Microbiol. 2008, 6, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Roe, A.J.; Yull, H.; Naylor, S.W.; Woodward, M.J.; Smith, D.G.E.; Gally, D.L. Heterogeneous surface expression of EspA translocon filaments by Escherichia coli O157:H7 is controlled at the posttranscriptional level. Infect. Immun. 2003, 71, 5900–5909. [Google Scholar] [CrossRef] [Green Version]
- Roe, A.J.; Tysall, L.; Dransfield, T.; Wang, D.; Fraser-Pitt, D.; Mahajan, A.; Constandinou, C.; Inglis, N.; Downing, A.; Talbot, R.; et al. Analysis of the expression, regulation and export of NleA-E in Escherichia coli O157:H7. Microbiology 2007, 153, 1350–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, X.; Oswald, E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J. Bacteriol. 2004, 186, 5486–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, H.J.; Pearson, J.S.; Badea, L.; Kelly, M.; Lucas, M.; Holloway, G.; Wagstaff, K.M.; Dunstone, M.A.; Sloan, J.; Whisstock, J.C.; et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 2010, 6, e1000898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Angulo, V.A.; Martínez-Santos, V.I.; Villaseñor, T.; Santana, F.J.; Huerta-Saquero, A.; Martínez, L.C.; Jiménez, R.; Lara-Ochoa, C.; Téllez-Sosa, J.; Bustamante, V.H.; et al. A distinct regulatory sequence is essential for the expression of a subset of nle genes in attaching and effacing Escherichia coli. J. Bacteriol. 2012, 194, 5589–5603. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Yu, H.B.; De Hoog, C.L.; Stoynov, N.; Li, Y.; Foster, L.J.; Finlay, B.B. Quantitative proteomic analysis of type III secretome of enteropathogenic Escherichia coli reveals an expanded effector repertoire for attaching/effacing bacterial pathogens. Mol. Cell. Proteom. 2012, 11, 692–709. [Google Scholar] [CrossRef] [Green Version]
- Ellermeier, C.D.; Ellermeier, J.R.; Slauch, J.M. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2005, 57, 691–705. [Google Scholar] [CrossRef]
- Schechter, L.M.; Jain, S.; Akbar, S.; Lee, C.A. The small nucleoid-binding proteins H-NS, HU, and Fis affect hilA expression in Salmonella enterica serovar Typhimurium. Infect. Immun. 2003, 71, 5432–5435. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, R.C.; Knodler, L.A.; Barhoumi, R.; Ross Payne, H.; Wu, J.; Gomez, G.; Pugh, R.; Lawhon, S.D.; Bäumler, A.J.; Steele-Mortimer, O.; et al. Spatial segregation of virulence gene expression during acute enteric infection with Salmonella enterica serovar Typhimurium. MBio 2014, 5, e00946-13. [Google Scholar] [CrossRef] [Green Version]
- Mouslim, C.; Delgado, M.; Groisman, E.A. Activation of the RcsC/YojN/RcsB phosphorelay system attenuates Salmonella virulence. Mol. Microbiol. 2004, 54, 386–395. [Google Scholar] [CrossRef]
- Golubeva, Y.A.; Sadik, A.Y.; Ellermeier, J.R.; Slauch, J.M. Integrating global regulatory input into the Salmonella pathogenicity Island 1 type III secretion system. Genetics 2012, 190, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.D.; Kim, K.; Slauch, J.M. PhoP-mediated repression of the SPI1 T3SS in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2019, 201, e00264-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prost, L.R.; Miller, S.I. The Salmonellae PhoQ sensor: Mechanisms of detection of phagosome signals. Cell. Microbiol. 2008, 10, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Groisman, E.A. Howard Hughes Medical Institute The PhoQ/PhoP regulatory network of Salmonella enterica. Adv. Exp. Med. Biol. 2008, 631, 7–21. [Google Scholar] [PubMed]
- Bijlsma, J.J.E.; Groisman, E.A. The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica. Mol. Microbiol. 2005, 57, 85–96. [Google Scholar] [CrossRef]
- Baisón-Olmo, F.; Galindo-Moreno, M.; Ramos-Morales, F. Host cell type-dependent translocation and PhoP-mediated positive regulation of the effector SseK1 of Salmonella enterica. Front. Microbiol. 2015, 6, 396. [Google Scholar] [CrossRef]
- Günster, R.A.; Matthews, S.A.; Holden, D.W.; Thurston, T.L.M. SseK1 and SseK3 T3SS effectors inhibit NF-κB signalling and necroptotic cell death in Salmonella-infected macrophages. Infect. Immun. 2017. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Soderholm, A.; Lung, T.W.F.; Giogha, C.; Hill, M.M.; Brown, N.F.; Hartland, E.; Teasdale, R.D. SseK3 Is a Salmonella effector that binds TRIM32 and modulates the host’s NF-κB signalling activity. PLoS ONE 2015, 10, e0138529. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.G.; Lomako, J.; Lomako, W.M.; Whelan, W.J.; Meyer, H.E.; Serwe, M.; Metzger, J.W. beta-Glucosylarginine: A new glucose-protein bond in a self-glucosylating protein from sweet corn. FEBS Lett. 1995, 376, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, L.; Yao, Q.; Li, L.; Dong, N.; Rong, J.; Gao, W.; Ding, X.; Sun, L.; Chen, X.; et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 2013, 501, 242–246. [Google Scholar] [CrossRef]
- Pearson, J.S.; Giogha, C.; Ong, S.Y.; Kennedy, C.L.; Kelly, M.; Robinson, K.S.; Lung, T.W.F.; Mansell, A.; Riedmaier, P.; Oates, C.V.L.; et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 2013, 501, 247–251. [Google Scholar] [CrossRef]
- Gao, X.; Pham, T.H.; Feuerbacher, L.A.; Chen, K.; Hays, M.P.; Singh, G.; Rueter, C.; Hurtado-Guerrero, R.; Hardwidge, P.R. Citrobacter rodentium NleB protein inhibits Tumor Necrosis Factor (TNF) receptor-associated factor 3 (TRAF3) ubiquitination to reduce host type I interferon production. J. Biol. Chem. 2016, 291, 18232–18238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Qaidi, S.; Chen, K.; Halim, A.; Siukstaite, L.; Rueter, C.; Hurtado-Guerrero, R.; Clausen, H.; Hardwidge, P.R. NleB/SseK effectors from Citrobacter rodentium, Escherichia coli, and Salmonella enterica display distinct differences in host substrate specificity. J. Biol. Chem. 2017, 292, 11423–11430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, D.; Günster, R.A.; Martino, L.; El Omari, K.; Wagner, A.; Thurston, T.L.M.; Rittinger, K. Structural basis for the glycosyltransferase activity of the Salmonella effector SseK3. J. Biol. Chem. 2018, 293, 5064–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.B.; Kim, Y.H.; Yoo, Y.; Kim, J.; Jun, S.-H.; Cho, J.W.; El Qaidi, S.; Walpole, S.; Monaco, S.; García-García, A.A.; et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat. Commun. 2018, 9, 4283. [Google Scholar] [CrossRef]
- Ding, J.; Pan, X.; Du, L.; Yao, Q.; Xue, J.; Yao, H.; Wang, D.-C.; Li, S.; Shao, F. Structural and functional insights into host death domains inactivation by the bacterial arginine GlcNAcyltransferase effector. Mol. Cell 2019, 74, 922–935.e6. [Google Scholar] [CrossRef] [PubMed]
- Newson, J.P.; Scott, N.E.; Yeuk Wah Chung, I.; Wong Fok Lung, T.; Giogha, C.; Gan, J.; Wang, N.; Strugnell, R.; Brown, N.F.; Cygler, M.; et al. Salmonella effectors SseK1 and SseK3 target death domain proteins in the TNF and TRAIL signaling pathways. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Li, S.; Li, X.; Shao, F.; Liu, L.; Hu, H.-G. Synthesis of and specific antibody generation for glycopeptides with arginine N-GlcNAcylation. Angew. Chem. Int. Ed. Engl. 2014, 53, 14517–14521. [Google Scholar] [CrossRef]
- Park, J.B.; Yoo, Y.; Cho, H.-S. Structural insights showing how arginine is able to be glycosylated by pathogenic effector proteins. BMB Rep. 2018, 51, 609–610. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; O’Connor, T.J. Beyond paralogs: The multiple layers of redundancy in bacterial pathogenesis. Front. Cell. Infect. Microbiol. 2017, 7, 467. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, X.; Pham, T.H.; Feuerbacher, L.A.; Lubos, M.-L.; Huang, M.; Olsen, R.; Mushegian, A.; Slawson, C.; Hardwidge, P.R. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 2013, 13, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Wong Fok Lung, T.; Giogha, C.; Creuzburg, K.; Ong, S.Y.; Pollock, G.L.; Zhang, Y.; Fung, K.Y.; Pearson, J.S.; Hartland, E.L. Mutagenesis and functional analysis of the bacterial arginine glycosyltransferase effector NleB1 from enteropathogenic Escherichia coli. Infect. Immun. 2016, 84, 1346–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornick, N.A.; Pitzer, J.; Helgerson, A.F.; Madsen, M.L.; Kurth, K.T.; Xiao, Q.; Minion, F.C. Use of signature-tagged mutagenesis to identify genes associated with colonization of sheep by E. coli O157:H7. Vet. Microbiol. 2017, 201, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misyurina, O.; Asper, D.J.; Deng, W.; Finlay, B.B.; Rogan, D.; Potter, A.A. The role of Tir, EspA, and NleB in the colonization of cattle by Shiga toxin producing Escherichia coli O26:H11. Can. J. Microbiol. 2010, 56, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Afset, J.E.; Bruant, G.; Brousseau, R.; Harel, J.; Anderssen, E.; Bevanger, L.; Bergh, K. Identification of virulence genes linked with diarrhea due to atypical enteropathogenic Escherichia coli by DNA microarray analysis and PCR. J. Clin. Microbiol. 2006, 44, 3703–3711. [Google Scholar] [CrossRef] [Green Version]
- Bugarel, M.; Martin, A.; Fach, P.; Beutin, L. Virulence gene profiling of enterohemorrhagic (EHEC) and enteropathogenic (EPEC) Escherichia coli strains: A basis for molecular risk assessment of typical and atypical EPEC strains. BMC Microbiol. 2011, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Baines, D.; Sumarah, M.; Kuldau, G.; Juba, J.; Mazza, A.; Masson, L. Aflatoxin, fumonisin and Shiga toxin-producing Escherichia coli infections in calves and the effectiveness of celmanax®/dairyman’s choiceTM applications to eliminate morbidity and mortality losses. Toxins 2013, 5, 1872–1895. [Google Scholar] [CrossRef]
- Bugarel, M.; Beutin, L.; Fach, P. Low-density macroarray targeting non-locus of enterocyte effacement effectors (nle genes) and major virulence factors of Shiga toxin-producing Escherichia coli (STEC): A new approach for molecular risk assessment of STEC isolates. Appl. Environ. Microbiol. 2010, 76, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Wickham, M.E.; Lupp, C.; Mascarenhas, M.; Vázquez, A.; Coombes, B.K.; Brown, N.F.; Coburn, B.A.; Deng, W.; Puente, J.L.; Karmali, M.A.; et al. Bacterial genetic determinants of non-O157 STEC outbreaks and hemolytic-uremic syndrome after infection. J. Infect. Dis. 2006, 194, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Buvens, G.; Piérard, D. Virulence profiling and disease association of verocytotoxin-producing Escherichia coli O157 and non-O157 isolates in Belgium. Foodborne Pathog. Dis. 2012, 9, 530–535. [Google Scholar] [CrossRef]
- de Boer, R.F.; Ferdous, M.; Ott, A.; Scheper, H.R.; Wisselink, G.J.; Heck, M.E.; Rossen, J.W.; Kooistra-Smid, A.M.D. Assessing the public health risk of Shiga toxin-producing Escherichia coli by use of a rapid diagnostic screening algorithm. J. Clin. Microbiol. 2015, 53, 1588–1598. [Google Scholar] [CrossRef] [Green Version]
- Ferdous, M.; Friedrich, A.W.; Grundmann, H.; de Boer, R.F.; Croughs, P.D.; Islam, M.A.; Kluytmans-van den Bergh, M.F.Q.; Kooistra-Smid, A.M.D.; Rossen, J.W.A. Molecular characterization and phylogeny of Shiga toxin–producing Escherichia coli isolates obtained from two Dutch regions using whole genome sequencing. Clin. Microbiol. Infect. 2016, 22, 642.e1–642.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matussek, A.; Jernberg, C.; Einemo, I.-M.; Monecke, S.; Ehricht, R.; Engelmann, I.; Löfgren, S.; Mernelius, S. Genetic makeup of Shiga toxin-producing Escherichia coli in relation to clinical symptoms and duration of shedding: A microarray analysis of isolates from Swedish children. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.A.; Dos Santos, L.F.; Dias, R.C.B.; Camargo, C.H.; Pinheiro, S.R.S.; Gomes, T.A.T.; Hernandes, R.T. Atypical enteropathogenic Escherichia coli as aetiologic agents of sporadic and outbreak-associated diarrhoea in Brazil. J. Med. Microbiol. 2016, 65, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bai, X.; Jin, Y.; Hu, B.; Wang, H.; Sun, H.; Fan, R.; Fu, S.; Xiong, Y. High prevalence of virulence genes in specific genotypes of atypical enteropathogenic Escherichia coli. Front. Cell. Infect. Microbiol. 2017, 7, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckner, M.M.C.; Croxen, M. a; Arena, E.T.; Finlay, B.B. A comprehensive study of the contribution of Salmonella enterica serovar Typhimurium SPI2 effectors to bacterial colonization, survival, and replication in typhoid fever, macrophage, and epithelial cell infection models. Virulence 2011, 2, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yu, C.; Ding, K.; Zhang, C.; Liao, C.; Jia, Y.; Li, J.; Cheng, X. Role of the sseK1 gene in the pathogenicity of Salmonella enterica serovar Enteritidis in vitro and in vivo. Microb. Pathog. 2018, 117, 270–275. [Google Scholar] [CrossRef]
- Zhang, X.; He, L.; Zhang, C.; Yu, C.; Yang, Y.; Jia, Y.; Cheng, X.; Li, Y.; Liao, C.; Li, J.; et al. The impact of sseK2 deletion on Salmonella enterica serovar Typhimurium virulence in vivo and in vitro. BMC Microbiol. 2019, 19, 182. [Google Scholar] [CrossRef]
- Lawley, T.D.; Chan, K.; Thompson, L.J.; Kim, C.C.; Govoni, G.R.; Monack, D.M. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006, 2, e11. [Google Scholar] [CrossRef] [Green Version]
- Nadler, C.; Baruch, K.; Kobi, S.; Mills, E.; Haviv, G.; Farago, M.; Alkalay, I.; Bartfeld, S.; Meyer, T.F.; Ben-Neriah, Y.; et al. The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog. 2010, 6, e1000743. [Google Scholar] [CrossRef] [Green Version]
- Ruchaud-Sparagano, M.-H.; Mühlen, S.; Dean, P.; Kenny, B. The enteropathogenic E. coli (EPEC) Tir effector inhibits NF-κB activity by targeting TNFα receptor-associated factors. PLoS Pathog. 2011, 7, e1002414. [Google Scholar] [CrossRef]
- Molloy, S. Bacterial pathogenesis: A sweet interaction with death for EPEC. Nat. Rev. Microbiol. 2013, 11, 659. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.S.; Hartland, E.L. A surprising sweetener from enteropathogenic Escherichia coli. Gut Microbes 2014, 5, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, N.E.; Giogha, C.; Pollock, G.L.; Kennedy, C.L.; Webb, A.I.; Williamson, N.A.; Pearson, J.S.; Hartland, E.L. The bacterial arginine glycosyltransferase effector NleB preferentially modifies Fas-associated death domain protein (FADD). J. Biol. Chem. 2017, 292, 17337–17350. [Google Scholar] [CrossRef] [Green Version]
- El Qaidi, S.; Zhu, C.; McDonald, P.; Roy, A.; Maity, P.K.; Rane, D.; Perera, C.; Hardwidge, P.R. High-throughput screening for bacterial glycosyltransferase inhibitors. Front. Cell. Infect. Microbiol. 2018, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, X.; Zha, H.; Fan, S.; Zhang, D.; Li, S.; Xiao, W. A pathogen-derived effector modulates host glucose metabolism by arginine GlcNAcylation of HIF-1α protein. PLoS Pathog. 2018, 14, e1007259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasche, S.; Arens, S.; Ceol, A.; Siszler, G.; Schmid, M.A.; Häuser, R.; Schwarz, F.; Wuchty, S.; Aloy, P.; Uetz, P.; et al. The EHEC-host interactome reveals novel targets for the translocated intimin receptor. Sci. Rep. 2014, 4, 7531. [Google Scholar] [CrossRef]
- Law, R.J.; Law, H.T.; Scurll, J.M.; Scholz, R.; Santos, A.S.; Shames, S.R.; Deng, W.; Croxen, M.A.; Li, Y.; de Hoog, C.L.; et al. Quantitative mass spectrometry identifies novel host binding partners for pathogenic Escherichia coli type III secretion system effectors. J. Proteome Res. 2016, 15, 1613–1622. [Google Scholar] [CrossRef]
- El Qaidi, S.; Scott, N.E.; Hays, M.P.; Geisbrecht, B.V.; Watkins, S.; Hardwidge, P.R. An intra-bacterial activity for a T3SS effector. Sci. Rep. 2020, 10, 1073. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Mechanism | ||||
|---|---|---|---|---|
| Inverting | Retaining | Unknown | ||
| Fold type | GT-A | 2 1, 7, 12, 13, 14, 16, 21, 29, 31, 42, 43, 82, 84, 109 | 6, 8, 15, 24, 27, 34, 44, 55, 62, 64, 78, 81, 88 | |
| GT-B | 1, 9, 10, 18, 19, 23, 28, 30, 33, 38, 41, 52, 63, 65, 68, 70, 80, 90, 104 | 3, 4, 5, 20, 35, 52, 72, 99, 107 | ||
| GT-C | 22, 39, 50, 53, 57, 58, 59, 66, 83, 85, 87 | |||
| Others | 26 (GT-E), 51 (lysozyme-type), | 101 (GT-D) | ||
| Unknown | 11, 17, 25, 37, 40, 47, 48, 49, 54, 56, 61, 67, 73, 74, 75, 76, 92, 94, 97, 98, 100, 102, 103, 105, 106, 108 | 32, 45, 60, 69, 71, 77, 79, 89, 93, 95, 96 | 91, 110 | |
| Organism. | Reference Strain | Effector Name | Protein Size (aa) | Genomic Context | References |
|---|---|---|---|---|---|
| C. rodentium | ICC168 | NleB | 329 | Genomic island GI4 | [106,107] |
| Enterohemorrhagic E. coli (EHEC) O157:H7 | EDL933 | NleB1 | 329 | O-island 122/SpLE3 | [106,108] |
| NleB2 | 326 | O-island 36/Phage CP-933K/Sp3 | |||
| Enteropathogenic E. coli (EPEC) O127:H6 | E2348/69 | NleB1 | 329 | Integrative element IE6 | [109,110] |
| NleB2 | 326 | Phage PP4 | |||
| S. enterica serovar Typhimurium | 14028 | SseK1 | 336 | [111,112,113] | |
| SseK2 | 348 | ||||
| SseK3 | 335 | Phage ST64B |
| NleB C. rodentium | NleB1 EPEC | NleB1 EHEC | NleB E. albertii | NleB E. marmotae | NleB2 | SseK1 | SseK2 | SseK3 | |
|---|---|---|---|---|---|---|---|---|---|
| NleB C. rodentium | |||||||||
| NleB1 EPEC | 88.75 | ||||||||
| NleB1 EHEC | 89.06 | 97.87 | |||||||
| NleB1 E. albertii | 88.45 | 97.57 | 99.09 | ||||||
| NleB E. marmotae | 71.42 | 71.73 | 71.73 | 71.43 | |||||
| NleB2 EPEC/EHEC | 59.82 | 61.04 | 61.35 | 60.43 | 60.43 | ||||
| SseK1 S. enterica | 57.45 | 58.05 | 59.57 | 58.97 | 58.97 | 53.68 | |||
| SseK2 S. enterica | 52.58 | 52.28 | 53.19 | 52.58 | 51.06 | 46.93 | 55.36 | ||
| SseK3 S. enterica | 51.98 | 53.19 | 53.50 | 52.89 | 54.10 | 47.55 | 54.93 | 75.22 | |
| NleB Y. hibernica | 32.52 | 32.83 | 34.04 | 34.04 | 34.04 | 33.13 | 35.12 | 33.14 | 33.43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araujo-Garrido, J.L.; Bernal-Bayard, J.; Ramos-Morales, F. Type III Secretion Effectors with Arginine N-Glycosyltransferase Activity. Microorganisms 2020, 8, 357. https://doi.org/10.3390/microorganisms8030357
Araujo-Garrido JL, Bernal-Bayard J, Ramos-Morales F. Type III Secretion Effectors with Arginine N-Glycosyltransferase Activity. Microorganisms. 2020; 8(3):357. https://doi.org/10.3390/microorganisms8030357
Chicago/Turabian StyleAraujo-Garrido, Juan Luis, Joaquín Bernal-Bayard, and Francisco Ramos-Morales. 2020. "Type III Secretion Effectors with Arginine N-Glycosyltransferase Activity" Microorganisms 8, no. 3: 357. https://doi.org/10.3390/microorganisms8030357