The Saltpan Microbiome Is Structured by Sediment Depth and Minimally Influenced by Variable Hydration



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Soil Physico-Chemical Analysis
2.3. DNA Extraction, 16S rRNA Gene Amplification, and Sequencing
2.4. Data Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Escapa, M.; Perillo, G.M.E.; Iribarne, O. Biogeomorphically driven salt pan formation in Sarcocornia-dominated salt-marshes. Geomorphology 2015, 228, 147–157. [Google Scholar] [CrossRef]
- Linhoss, A.C.; Underwood, W. V Modeling salt panne land-cover suitability under sea-level rise. J. Coast. Res. 2015, 32, 1116–1125. [Google Scholar] [CrossRef]
- Lowenstein, T.K.; Hardie, L.A. Criteria for the recognition of salt-pan evaporites. Sedimentology 1985, 32, 627–644. [Google Scholar] [CrossRef]
- Wilms, R.; Sass, H.; Köpke, B.; Köster, J.; Cypionka, H.; Engelen, B. Specific bacterial, archaeal, and eukaryotic communities in tidal-flat sediments along a vertical profile of several meters. Appl. Environ. Microbiol. 2006, 72, 2756–2764. [Google Scholar] [CrossRef] [Green Version]
- Gao, S. Geomorphology and sedimentology of tidal flats. In Coastal Wetlands. An Integrated Ecosystem Approach, 2nd ed.; Perillo, G., Wolanski, E., Cahoon, D., Hopkinson, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 359–381. [Google Scholar]
- Peterson, M.S.; Waggy, G.L.; Woodrey, M.S. Grand Bay national estuarine research reserve: An ecological characterization. Grand Bay National Estuarine Research Reserve 2007. [Google Scholar]
- Craft, C.; Clough, J.; Ehman, J.; Joye, S.; Park, R.; Pennings, S.; Guo, H.; Machmuller, M. Forecasting the effects of accelerated sea-level rise on tidal marsh ecosystem services. Front. Ecol. Environ. 2009, 7, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.S.; Oh, H.M.; Kang, H.; Park, S.S.; Chun, J. Remarkable bacterial diversity in the tidal flat sediment as revealed by 16S rDNA analysis. J. Microbiol. Biotechnol. 2004, 14, 205–211. [Google Scholar]
- Stevens, H.; Brinkhoff, T.; Rink, B.; Vollmers, J.; Simon, M. Diversity and abundance of Gram positive bacteria in a tidal flat ecosystem. Environ. Microbiol. 2007, 9, 1810–1822. [Google Scholar] [CrossRef]
- Zedler, J.B.; Kercher, S. Wetland resources: Status, trends, ecosystem services, and restorability. Annu. Rev. Environ. Resour. 2005, 30, 39–74. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Lee, S.H.; Kang, H. Denitrification rates and community structure of denitrifying bacteria in newly constructed wetland. Eur. J. Soil Biol. 2011, 47, 24–29. [Google Scholar] [CrossRef]
- Mitsch, W.J.; Mander, Ü. Wetlands and carbon revisited. Ecol. Eng. 2018, 114, 1–6. [Google Scholar] [CrossRef]
- Foulquier, A.; Volat, B.; Neyra, M.; Bornette, G.; Montuelle, B. Long-term impact of hydrological regime on structure and functions of microbial communities in riverine wetland sediments. FEMS Microbiol. Ecol. 2013, 85, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.S.; Haug, L.A.; Rivera, V.A.; Gonzalez, L.M.H.; Kelly, J.J.; Miller, W.M.; Wells, G.F.; Packman, A.I. Soil hydrology drives ecological niche differentiation in a native prairie microbiome. FEMS Microbiol. Ecol. 2020, 96, fiz163. [Google Scholar] [CrossRef]
- Szukics, U.; Abell, G.C.J.; Hödl, V.; Mitter, B.; Sessitsch, A.; Hackl, E.; Zechmeister-Boltenstern, S. Nitrifiers and denitrifiers respond rapidly to changed moisture and increasing temperature in a pristine forest soil. FEMS Microbiol. Ecol. 2010, 72, 395–406. [Google Scholar] [CrossRef]
- Evans, S.E.; Wallenstein, M.D.; Burke, I.C. Is bacterial moisture niche a good predictor of shifts in community composition under long-term drought? Ecology 2014, 95, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, B.M.; Moroenyane, I.; Sherman, C.; Lee, Y.K.; Adams, J.M.; Steinberger, Y. Trends in taxonomic and functional composition of soil microbiome along a precipitation gradient in Israel. Microb. Ecol. 2017, 74, 168–176. [Google Scholar] [CrossRef]
- Kleyer, H.; Tecon, R.; Or, D. Rapid shifts in bacterial community assembly under static and dynamic hydration conditions in porous media. Appl. Environ. Microbiol. 2019, 86, e02057-19. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Lee, S.-H.; Mitsch, W.J.; Kang, H. Different responses of denitrification rates and denitrifying bacterial communities to hydrologic pulsing in created wetlands. Soil Biol. Biochem. 2010, 42, 1721–1727. [Google Scholar] [CrossRef]
- Brown, D.P.; Latto, A.; Berg, R. Tropical Storm Gordon (AL072018); National Hurricane Center: Miami, FL, USA, 2019. [Google Scholar]
- He, Y.; DeSutter, T.; Hopkins, D.; Jia, X.; Wysocki, D.A. Predicting ECe of the saturated paste extract from value of EC1: 5. Can. J. Soil Sci. 2013, 93, 585–594. [Google Scholar] [CrossRef]
- Simón, M.; Cabezas, O.; García, I.; Martínez, P. A new method for the estimation of total dissolved salts in saturation extracts of soils from electrical conductivity. Eur. J. Soil Sci. 1994, 45, 153–157. [Google Scholar] [CrossRef]
- Kettler, T.A.; Doran, J.W.; Gilbert, T.L. Simplified method for soil particle-size determination to accompany soil-quality analyses. Soil Sci. Soc. Am. J. 2001, 65, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, B.W.G.; Jackson, C.R. Biogeographic Patterns Between Bacterial Phyllosphere Communities of the Southern Magnolia (Magnolia grandiflora) in a Small Forest. Microb. Ecol. 2016, 71, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the Effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-Based Studies. PLoS One 2011, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Maidak, B.L.; Cole, J.R.; Lilburn, T.G.; Parker Jr, C.T.; Saxman, P.R.; Stredwick, J.M.; Garrity, G.M.; Li, B.; Olsen, G.J.; Pramanik, S. The RDP (ribosomal database project) continues. Nucleic Acids Res. 2000, 28, 173–174. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The vegan package. Community Ecol. Packag. 2007, 10, 631–637. [Google Scholar]
- Paulson, J.N.; Pop, M.; Bravo, H.C. Metastats: An improved statistical method for analysis of metagenomic data. Genome Biol. 2011, 12, P17. [Google Scholar] [CrossRef] [Green Version]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2013, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poret-Peterson, A.T.; Albu, S.; McClean, A.E.; Kluepfel, D.A. Shifts in soil bacterial communities as a function of carbon source used during anaerobic soil disinfestation. Front. Environ. Sci. 2018, 6, 160. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Diniz-Filho, J.A.F.; Soares, T.N.; Lima, J.S.; Dobrovolski, R.; Landeiro, V.L.; Telles, M.P.; de Campos Telles, M.P.; Rangel, T.F.; Bini, L.M. Mantel test in population genetics. Genet. Mol. Biol. 2013, 36, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Osland, M.J.; Enwright, N.M.; Day, R.H.; Gabler, C.A.; Stagg, C.L.; Grace, J.B. Beyond just sea-level rise: Considering macroclimatic drivers within coastal wetland vulnerability assessments to climate change. Glob. Chang. Biol. 2016, 22, 1–11. [Google Scholar] [CrossRef]
- Wu, W.; Yeager, K.M.; Peterson, M.S.; Fulford, R.S. Neutral models as a way to evaluate the Sea Level Affecting Marshes Model (SLAMM). Ecol. Modell. 2015, 303, 55–69. [Google Scholar] [CrossRef]
- Bowen, J.L.; Morrison, H.G.; Hobbie, J.E.; Sogin, M.L. Salt marsh sediment diversity: A test of the variability of the rare biosphere among environmental replicates. ISME J. 2012, 6, 2014–2023. [Google Scholar] [CrossRef] [Green Version]
- Dini-Andreote, F.; e Silva, M.d.C.P.; Triado-Margarit, X.; Casamayor, E.O.; Van Elsas, J.D.; Salles, J.F. Dynamics of bacterial community succession in a salt marsh chronosequence: Evidences for temporal niche partitioning. ISME J. 2014, 8, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Ma, B.; Yu, J.; Chang, S.X.; Xu, J.; Li, Y.; Wang, G.; Han, G.; Bo, G.; Chu, X. Bacterial community structure and function shift along a successional series of tidal flats in the Yellow River Delta. Sci. Rep. 2016, 6, 36550. [Google Scholar] [CrossRef]
- Bernardet, J.F.; Bowman, J.P. The genus flavobacterium. The prokaryotes 2006, 7, 481–531. [Google Scholar]
- Lee, E.; Shin, D.; Hyun, S.P.; Ko, K.; Moon, H.S.; Koh, D.; Ha, K.; Kim, B. Periodic change in coastal microbial community structure associated with submarine groundwater discharge and tidal fluctuation. Limnol. Oceanogr. 2017, 62, 437–451. [Google Scholar] [CrossRef]
- Cheung, M.K.; Wong, C.K.; Chu, K.H.; Kwan, H.S. Community structure, dynamics and interactions of bacteria, Archaea and fungi in subtropical coastal wetland sediments. Sci. Rep. 2018, 8, 14397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behera, P.; Mahapatra, S.; Mohapatra, M.; Kim, J.Y.; Adhya, T.K.; Raina, V.; Suar, M.; Pattnaik, A.K.; Rastogi, G. Salinity and macrophyte drive the biogeography of the sedimentary bacterial communities in a brackish water tropical coastal lagoon. Sci. Total Environ. 2017, 595, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Holmström, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Cho, J.C.; Giovannoni, S.J. Parvularcula bermudensis gen. nov., sp. nov., a marine bacterium that forms a deep branch in the α-Proteobacteria. Int. J. Syst. Evol. Microbiol. 2003, 53, 1031–1036. [Google Scholar] [CrossRef]
- Koblížek, M.; Béjà, O.; Bidigare, R.R.; Christensen, S.; Benitez-Nelson, B.; Vetriani, C.; Kolber, M.K.; Falkowski, P.G.; Kolber, Z.S. Isolation and characterization of Erythrobacter sp. strains from the upper ocean. Arch. Microbiol. 2003, 180, 327–338. [Google Scholar] [CrossRef]
- Schlesner, H.; Rensmann, C.; Tindall, B.J.; Gade, D.; Rabus, R.; Pfeiffer, S.; Hirsch, P. Taxonomic heterogeneity within the Planctomycetales as derived by DNA–DNA hybridization, description of Rhodopirellula baltica gen. nov., sp. nov., transfer of Pirellula marina to the genus Blastopirellula gen. nov. as Blastopirellula marina comb. nov. and emended description of the genus Pirellula. Int. J. Syst. Evol. Microbiol. 2004, 54, 1567–1580. [Google Scholar]
- Yoon, J.H.; Kang, S.J.; Oh, T.K. Marinomonas dokdonensis sp. nov., isolated from sea water. Int. J. Syst. Evol. Microbiol. 2005, 55, 2303–2307. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, Y.; Kurahashi, M.; Sakiyama, Y.; Ohuchi, M.; Yokota, A.; Harayama, S. Phycisphaera mikurensis gen. nov., sp. nov., isolated from a marine alga, and proposal of Phycisphaeraceae fam. nov., Phycisphaerales ord. nov. and Phycisphaerae classis nov. in the phylum Planctomycetes. J. Gen. Appl. Microbiol. 2009, 55, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Quail, Z.X.; Zeng, D.N.; Xiao, Y.P.; Roh, S.W.; Nam, Y.D.; Chang, H.W.; Yoon, J.H.; Oh, H.M.; Bae, J.W. Henriciella marina gen. nov., sp. nov., a novel member of the family Hyphomonadaceae isolated from the East Sea. J. Microbiol. 2009, 47, 156–161. [Google Scholar] [CrossRef]
- Chen, C.; Zheng, Q.; Wang, Y.N.; Yan, X.J.; Hao, L.K.; Du, X.; Jiao, N. Stakelama pacifica gen. nov., sp. nov., a new member of the family Sphingomonadaceae isolated from the Pacific Ocean. Int. J. Syst. Evol. Microbiol. 2010, 60, 2857–2861. [Google Scholar] [CrossRef] [PubMed]
- Interaminense, J.A.; Nascimento, D.C.O.; Ventura, R.F.; Batista, J.E.C.; Souza, M.M.C.; Hazin, F.H.V.; Pontes-Filho, N.T.; Lima-Filho, J.V. Recovery and screening for antibiotic susceptibility of potential bacterial pathogens from the oral cavity of shark species involved in attacks on humans in Recife, Brazil. J. Med. Microbiol. 2010, 59, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.H.; Lee, S.D. Altererythrobacter marensis sp. nov., isolated from seawater. Int. J. Syst. Evol. Microbiol. 2010, 60, 307–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzetti, I.; Fuchs, B.M.; Gerdts, G.; Wichels, A.; Wiltshire, K.H.; Amann, R. Temporal variability of coastal Planctomycetes clades at Kabeltonne station, North Sea. Appl. Environ. Microbiol. 2011, 77, 5009–5017. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Zhou, Y.; Lai, Q.; Li, X.; Dong, P.; Yang, X.; Zhang, B.; Zhang, J.; Zheng, X.; Tian, Y. Sinobacterium caligoides gen. nov., sp. nov., a new member of the family Oceanospirillaceae isolated from the South China Sea, and emended description of Amphritea japonica. Int. J. Syst. Evol. Microbiol. 2013, 63, 2095–2100. [Google Scholar] [CrossRef]
- Li, N.; Williams, H.N. 454 Pyrosequencing reveals diversity of Bdellovibrio and like organisms in fresh and salt water. Antonie Van Leeuwenhoek 2015, 107, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Koval, S.F.; Williams, H.N.; Stine, O.C. Reclassification of Bacteriovorax marinus as Halobacteriovorax marinus gen. nov., comb. nov. and Bacteriovorax litoralis as Halobacteriovorax litoralis comb. nov.; description of Halobacteriovoraceae fam. nov. in the class Deltaproteobacteria. Int. J. Syst. Evol. Microbiol. 2015, 65, 305–311. [Google Scholar] [CrossRef]
- Lin, C.Y.; Zhang, X.Y.; Liu, A.; Liu, C.; Song, X.Y.; Su, H.N.; Qin, Q.L.; Xie, B.B.; Zhang, Y.Z. Marivirga atlantica sp. nov., isolated from seawater and emended description of the genus Marivirga. Int. J. Syst. Evol. Microbiol. 2015, 65, 1515–1519. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.T.; Lee, J.S.; Yoon, J.H. Gaetbulibacter aquiaggeris sp. nov., a member of the Flavobacteriaceae isolated from seawater. Int. J. Syst. Evol. Microbiol. 2016, 66, 1131–1137. [Google Scholar] [CrossRef]
- Simon, M.; Scheuner, C.; Meier-Kolthoff, J.P.; Brinkhoff, T.; Wagner-Döbler, I.; Ulbrich, M.; Klenk, H.-P.; Schomburg, D.; Petersen, J.; Göker, M. Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 2017, 11, 1483–1499. [Google Scholar] [CrossRef]
- Cho, J.C.; Giovannoni, S.J. Robiginitalea biformata gen. nov., sp. nov., a novel marine bacterium in the family Flavobacteriaceae with a higher G+ C content. Int. J. Syst. Evol. Microbiol. 2004, 54, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Lee, S.Y.; Oh, T.K. Gaetbulibacter lutimaris sp. nov., isolated from a tidal flat sediment. Int. J. Syst. Evol. Microbiol. 2013, 63, 995–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Choi, S.J.; Choi, J.; Yoon, J.H. Paraglaciecola aestuariivivens sp. nov., isolated from a tidal flat. Int. J. Syst. Evol. Microbiol. 2017, 67, 4754–4759. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Cha, I.T.; Kim, S.J.; Shin, K.S.; Hong, Y.; Roh, D.H.; Rhee, S.K. Salinisphaera orenii sp. nov., isolated from a solar saltern. Int. J. Syst. Evol. Microbiol. 2012, 62, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.J.; Lafay, B.; Christen, R.; Fernandez, L.; Acquaviva, M.; Bonin, P.; Bertrand, J.-C. Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int. J. Syst. Evol. Microbiol. 1992, 42, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFrank, J.J.; Beaudry, W.T.; Cheng, T.C.; Harvey, S.P.; Stroup, A.N.; Szafraniec, L.L. Screening of halophilic bacteria and Alteromonas species for organophosphorus hydrolyzing enzyme activity. Chem. Biol. Interact. 1993, 87, 141–148. [Google Scholar] [CrossRef]
- Zhong, Z.P.; Liu, Y.; Hou, T.T.; Liu, H.C.; Zhou, Y.G.; Wang, F.; Liu, Z.P. Marivita lacus sp. nov., isolated from a saline lake. Int. J. Syst. Evol. Microbiol. 2015, 65, 1889–1894. [Google Scholar] [CrossRef]
- Shah, H.N.; Collins, M.D. Proposal for reclassification of Bacteroides asaccharolyticus, Bacteroides gingivalis, and Bacteroides endodontalis in a new genus, Porphyromonas. Int. J. Syst. Evol. Microbiol. 1988, 38, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.L.; Shaw, J.G. Aeromonas spp. clinical microbiology and disease. J. Infect. 2011, 62, 109–118. [Google Scholar] [CrossRef]
- McSpadden Gardener, B.B. Ecology of Bacillus and Paenibacillus spp. in agricultural systems. Phytopathology 2004, 94, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Bruns, A.; Rohde, M.; Berthe-Corti, L. Muricauda ruestringensis gen. nov., sp. nov., a facultatively anaerobic, appendaged bacterium from German North Sea intertidal sediment. Int. J. Syst. Evol. Microbiol. 2001, 51, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choo, Y.J.; Song, J.; Lee, J.S.; Lee, K.C.; Cho, J.C. Marinobacterium litorale sp. nov. in the order Oceanospirillales. Int. J. Syst. Evol. Microbiol. 2007, 57, 1659–1662. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Kasai, H.; Matsuo, Y.; Ōmura, S.; Shizuri, Y.; Takahashi, Y. Ilumatobacter fluminis gen. nov., sp. nov., a novel actinobacterium isolated from the sediment of an estuary. J. Gen. Appl. Microbiol. 2009, 55, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekeman, B.; Kerckhof, F.; Cremers, G.; De Vos, P.; Vandamme, P.; Boon, N.; Op den Camp, H.J.M.; Heylen, K. New Methyloceanibacter diversity from North Sea sediments includes methanotroph containing solely the soluble methane monooxygenase. Environ. Microbiol. 2016, 18, 4523–4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.G. Mycobacterium tuberculosis: Here today, and here tomorrow. Nat. Rev. Mol. cell Biol. 2001, 2, 569–578. [Google Scholar] [CrossRef]
- Jiang, F.; Li, W.; Xiao, M.; Dai, J.; Kan, W.; Chen, L.; Li, W.; Fang, C.; Peng, F. Luteolibacter luojiensis sp. nov., isolated from Arctic tundra soil, and emended description of the genus Luteolibacter. Int. J. Syst. Evol. Microbiol. 2012, 62, 2259–2263. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Katayama, T.; Yamagishi, T.; Hanada, S.; Tamaki, H.; Kamagata, Y.; Oshima, K.; Hattori, M.; Marumo, K.; Nedachi, M. Methyloceanibacter caenitepidi gen. nov., sp. nov., a facultatively methylotrophic bacterium isolated from marine sediments near a hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2014, 64, 462–468. [Google Scholar] [CrossRef]
- Oren, A.; Arahal, D.R.; Ventosa, A. Emended descriptions of genera of the family Halobacteriaceae. Int. J. Syst. Evol. Microbiol. 2009, 59, 637–642. [Google Scholar] [CrossRef]
- Chen, S.; Liu, H.C.; Zhou, J.; Xiang, H. Haloparvum sedimenti gen. nov., sp. nov., a member of the family Haloferacaceae. Int. J. Syst. Evol. Microbiol. 2016, 66, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Tourna, M.; Stieglmeier, M.; Spang, A.; Könneke, M.; Schintlmeister, A.; Urich, T.; Engel, M.; Schloter, M.; Wagner, M.; Richter, A. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. USA 2011, 108, 8420–8425. [Google Scholar] [CrossRef] [Green Version]
- Iino, T.; Tamaki, H.; Tamazawa, S.; Ueno, Y.; Ohkuma, M.; Suzuki, K.; Igarashi, Y.; Haruta, S. Candidatus Methanogranum caenicola: A novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ. 2013, ME12189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, M. Microbiome Community Ecology: Fundamentals and Applications; Springer: Berlin, Germany, 2015; ISBN 3319116657. [Google Scholar]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minegishi, H.; Echigo, A.; Nagaoka, S.; Kamekura, M.; Usami, R. Halarchaeum acidiphilum gen. nov., sp. nov., a moderately acidophilic haloarchaeon isolated from commercial solar salt. Int. J. Syst. Evol. Microbiol. 2010, 60, 2513–2516. [Google Scholar] [CrossRef]
- Ivanova, N.; Rohde, C.; Munk, C.; Nolan, M.; Lucas, S.; Del Rio, T.G.; Tice, H.; Deshpande, S.; Cheng, J.F.; Tapia, R. Complete genome sequence of Truepera radiovictrix type strain (RQ-24 T). Stand. Genomic Sci. 2011, 4, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Song, J.; Yoshizawa, S.; Choi, A.; Cho, J.-C.; Kogure, K. Rubrivirga marina gen. nov., sp. nov., a member of the family Rhodothermaceae isolated from deep seawater. Int. J. Syst. Evol. Microbiol. 2013, 63, 2229–2233. [Google Scholar] [CrossRef] [Green Version]
- Sickel, W.; Kinkel, D.B.; Knief, C. Ex situ irrigation of Atacama soil stimulates bacterial respiration but does not induce changes in the microbial community. Proc. Geophys. Res. Abstr. 2019, 21. [Google Scholar]
- Hu, Y.; Wang, L.; Xi, X.; Hu, J.; Hou, Y.; Le, Y.; Fu, X. Effects of salinity on soil bacterial and archaeal community in estuarine wetlands and its implications for carbon sequestration: Verification in the Yellow River Delta. Chem. Ecol. 2016, 32, 669–683. [Google Scholar] [CrossRef]
- Franklin, R.B.; Morrissey, E.M.; Morina, J.C. Changes in abundance and community structure of nitrate-reducing bacteria along a salinity gradient in tidal wetlands. Pedobiologia (Jena). 2017, 60, 21–26. [Google Scholar] [CrossRef]
- Jiang, X.; Hou, L.; Zheng, Y.; Liu, M.; Yin, G.; Gao, J.; Li, X.; Wang, R.; Yu, C.; Lin, X. Salinity-driven shifts in the activity, diversity, and abundance of anammox bacteria of estuarine and coastal wetlands. Phys. Chem. Earth, Parts A/B/C 2017, 97, 46–53. [Google Scholar] [CrossRef]
- Chambers, L.G.; Reddy, K.R.; Osborne, T.Z. Short-term response of carbon cycling to salinity pulses in a freshwater wetland. Soil Sci. Soc. Am. J. 2011, 75, 2000–2007. [Google Scholar] [CrossRef]
- Neubauer, S.C.; Franklin, R.B.; Berrier, D.J. Saltwater intrusion into tidal freshwater marshes alters the biogeochemical processing of organic carbon. Biogeosciences 2013, 10, 8171–8183. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.D.; Dougill, A.J.; Elliott, D.R.; Mairs, H. Seasonal differences in soil CO2 efflux and carbon storage in Ntwetwe Pan, Makgadikgadi Basin, Botswana. Geoderma 2014, 219, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Emerson, D.; Bellows, W.; Keller, J.K.; Moyer, C.L.; Sutton-Grier, A.; Megonigal, J.P. Anaerobic metabolism in tidal freshwater wetlands: II. Effects of plant removal on archaeal microbial communities. Estuaries Coasts 2013, 36, 471–481. [Google Scholar] [CrossRef]
- Weston, N.B.; Vile, M.A.; Neubauer, S.C.; Velinsky, D.J. Accelerated microbial organic matter mineralization following salt-water intrusion into tidal freshwater marsh soils. Biogeochemistry 2011, 102, 135–151. [Google Scholar] [CrossRef]
- Steppe, T.F.; Paerl, H.W. Potential N2 fixation by sulfate-reducing bacteria in a marine intertidal microbial mat. Aquat. Microb. Ecol. 2002, 28, 1–12. [Google Scholar] [CrossRef]
- Bertics, V.J.; Löscher, C.R.; Salonen, I.; Dale, A.W.; Gier, J.; Schmitz, R.A.; Treude, T. Occurrence of benthic microbial nitrogen fixation coupled to sulfate reduction in the seasonally hypoxic Eckernförde Bay, Baltic Sea. Biogeosciences 2013, 10, 1243–1258. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Warry, F.Y.; Cook, P.L.M. The balance between nitrogen fixation and denitrification on vegetated and non-vegetated intertidal sediments. Limnol. Oceanogr. 2016, 61, 2058–2075. [Google Scholar] [CrossRef]
- Bae, H.S.; Morrison, E.; Chanton, J.P.; Ogram, A. Methanogens are major contributors to nitrogen fixation in soils of the Florida Everglades. Appl. Environ. Microbiol. 2018, 84, e02222-17. [Google Scholar] [CrossRef] [Green Version]

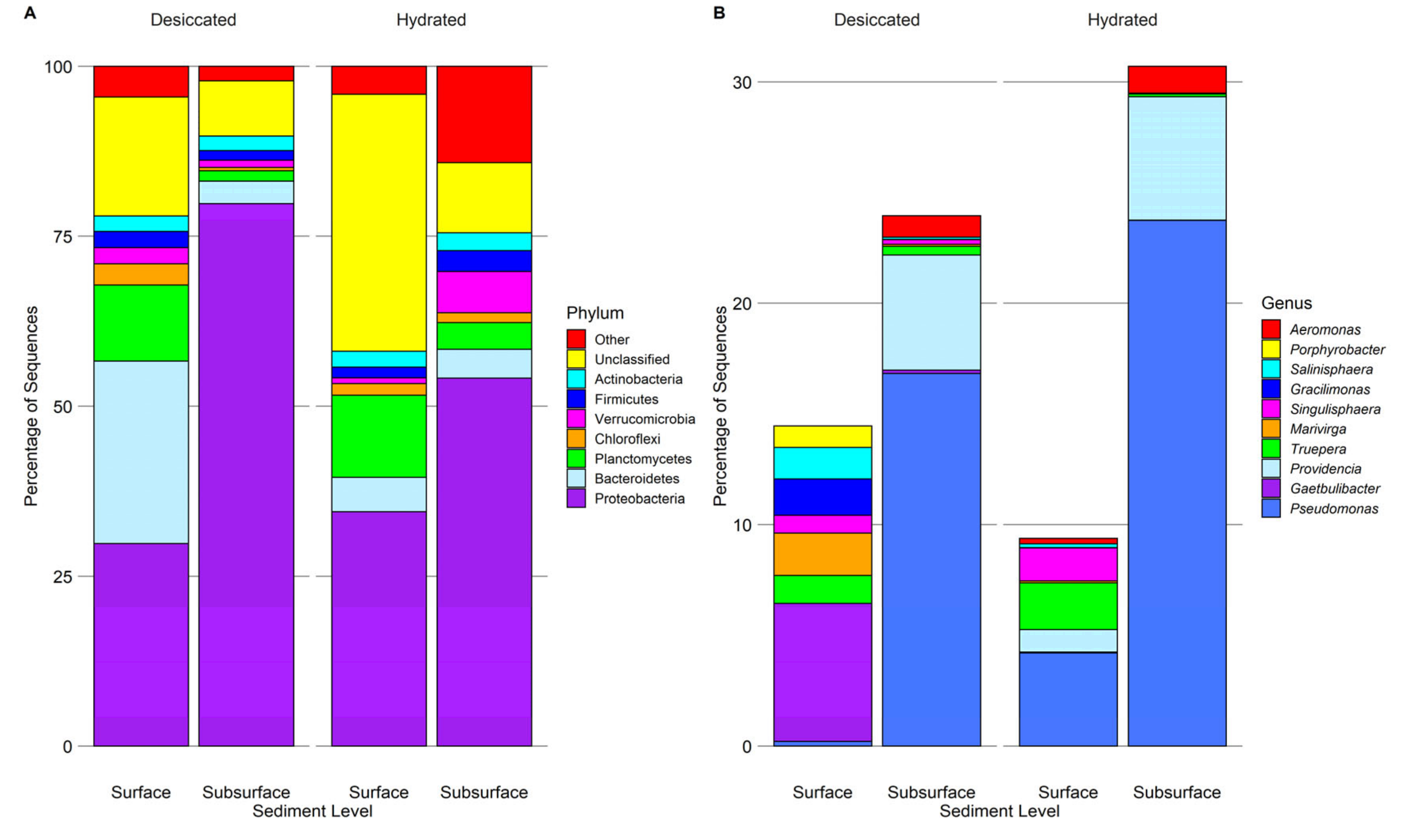

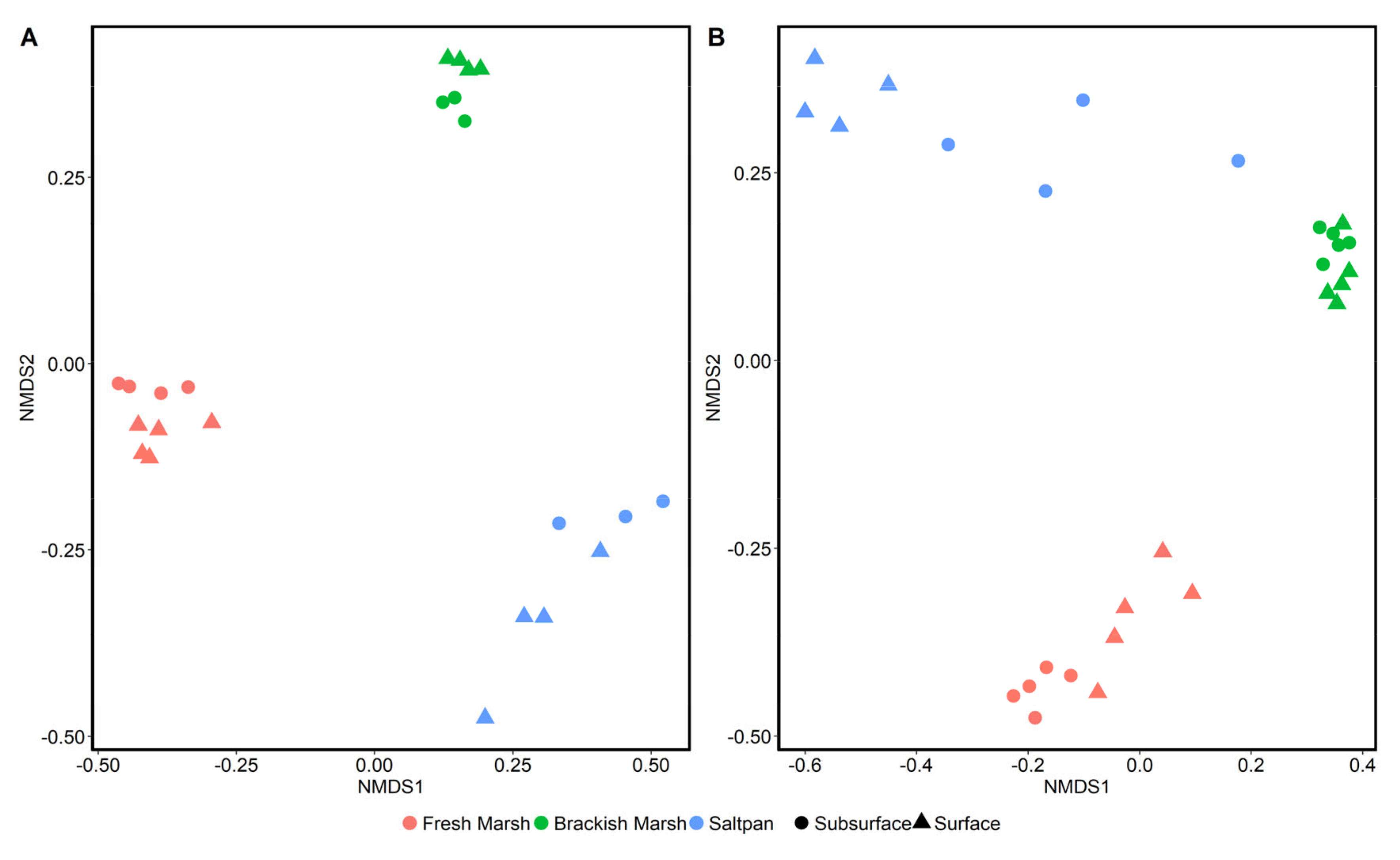

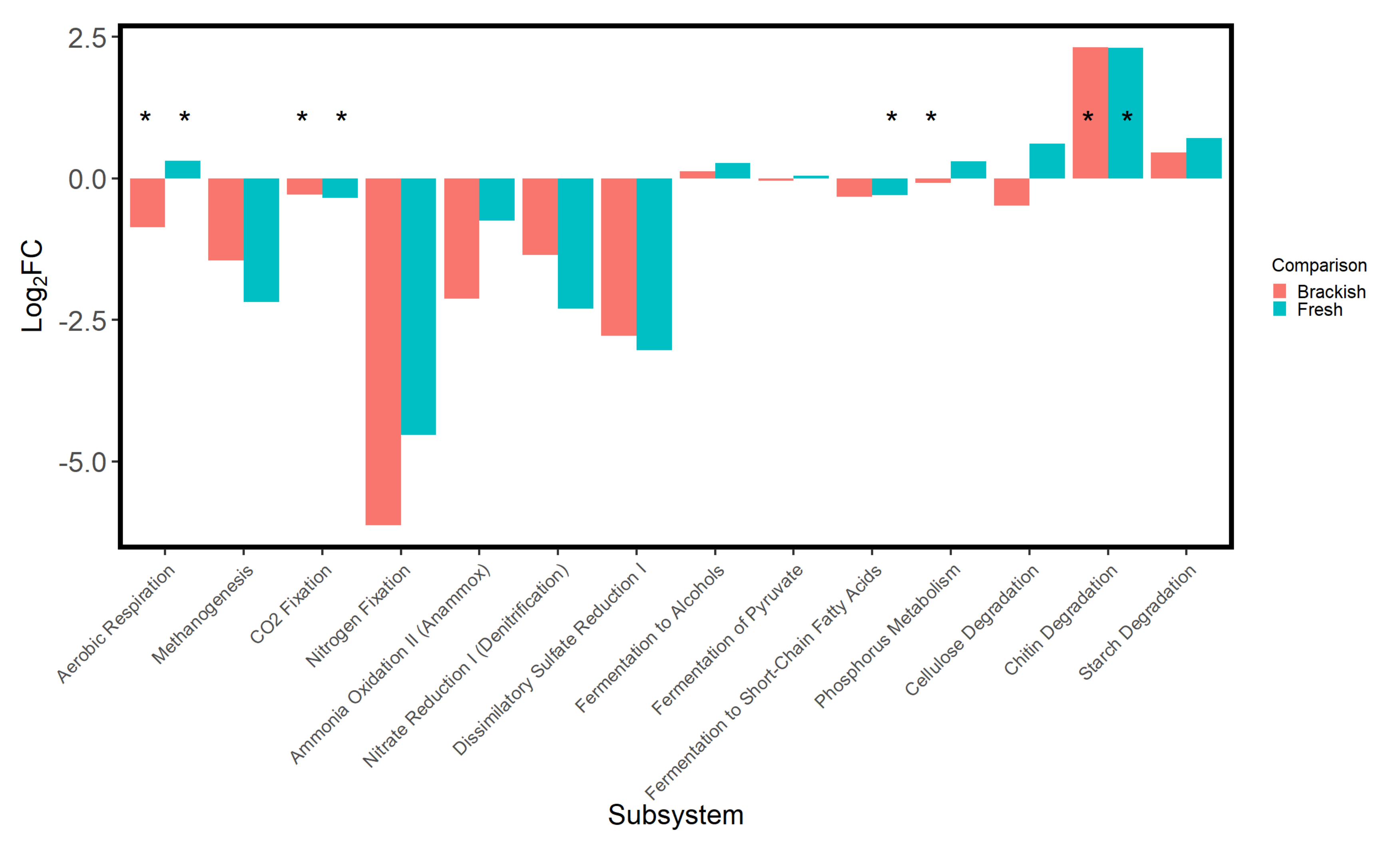

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Desiccated Surface | Hydrated Surface | Desiccated Subsurface | Hydrated Subsurface |
|---|---|---|---|---|
| Salinity (PSU) | 25.6 ± 0.13 | 14.7 ± 0.06 | 14.9 ± 0.03 | 13.6 ± 0.06 |
| pH | 5.91 ± 0.02 | 6.31 ± 0.11 | 6.33 ± 0.06 | 6.82 ± 0.04 |
| OM (%) | 5.53 ± 0.35 | 4.73 ± 0.03 | 4.08 ± 0.23 | 3.43 ± 0.22 |
| %Sand | 67.9 | 62.8 | 43.5 | 36.8 |
| %Silt | 28.7 | 35.1 | 49.8 | 56.9 |
| %Clay | 3.4 | 2.0 | 6.7 | 6.3 |
| Surface | Subsurface | ||||
|---|---|---|---|---|---|
| OTU | Identity | p-value | OTU | Identity | p-value |
| 15 | Truepera | 0.001 | 3 | Pseudomonas | 0.008 |
| 22 | Salinisphaera | 0.004 | 7 | Providencia | 0.004 |
| 43 | Marivita | 0.007 | 35 | Aeromonas | 0.007 |
| 91 | Alteromonas | 0.002 | 54 | Stenotrophomonas | 0.005 |
| 106 | Blastopirellula | 0.003 | 63 | Alcaligenes | 0.008 |
| 131 | Erythrobacter | 0.010 | 439 | Bacteroides | 0.002 |
| 138 | Halobacteriovorax | 0.007 | 444 | Bacillus | 0.010 |
| 170 | Henriciella | 0.002 | 674 | Paenibacillus | 0.007 |
| 171 | Aquisphaera | 0.005 | 741 | Bacteroides | 0.005 |
| 221 | Robiginitalea | 0.001 | |||
| 353 | Geodermatophilus | 0.003 | |||
| 367 | Pseudoalteromonas | 0.009 | |||
| 384 | Parvularcula | 0.003 | |||
| 548 | Vampirovibrio | 0.006 | |||
| 583 | Parvularcula | 0.007 | |||
| Desiccated | Hydrated | ||||
| OTU | Identity | p-value | OTU | Identity | p-value |
| 860 | Flavobacterium | 0.046 | 319 | Muricauda | 0.048 |
| 341 | Vampirovibrio | 0.046 | |||
| 346 | Mycobacterium | 0.020 | |||
| 389 | Luteolibacter | 0.021 | |||
| 404 | Marinobacterium | 0.041 | |||
| 435 | Aquisphaera | 0.025 | |||
| 529 | Methyloceanibacter | 0.020 | |||
| 590 | Marivirga | 0.038 | |||
| 688 | Ilumatobacter | 0.040 | |||
| OTU | Identity | Mean (Hydrated) | Mean (Desiccated) | p-value | Mean (Surface) | Mean (Subsurface) | p-value |
|---|---|---|---|---|---|---|---|
| 102 | Planctomycetaceae | 1.61 × 10−3 | 3.23 × 10−4 | 0.031 | 1.67 × 10−3 | 2.63 × 10−4 | 0.017 |
| 319 | Flavobacteriaceae | 3.18 × 10−4 | 0.00 | 0.048 | 3.18 × 10−4 | 0.00 | 0.047 |
| 341 | Bdellovibrionaceae | 3.66 × 10−4 | 0.00 | 0.046 | 3.66 × 10−4 | 0.00 | 0.046 |
| 342 | Alphaproteobacteria (c) | 3.04 × 10−4 | 0.00 | 0.044 | 3.04 × 10−4 | 0.00 | 0.044 |
| 403 | Alphaproteobacteria (c) | 2.05 × 10−4 | 4.90 × 10−5 | 0.035 | 2.04 × 10−4 | 5.00 × 10−5 | 0.043 |
| 404 | Oceanospirillaceae | 2.57 × 10−4 | 0.00 | 0.041 | 2.57 × 10−4 | 0.00 | 0.041 |
| 470 | Unclassified | 1.62 × 10−4 | 0.00 | 0.049 | 1.62 × 10−4 | 0.00 | 0.049 |
| 571 | Rhodospirillales | 1.42 × 10−4 | 0.00 | 0.04 | 1.42 × 10−4 | 0.00 | 0.04 |
| 590 | Flammeovirgaceae | 1.35 × 10−4 | 0.00 | 0.038 | 1.35 × 10−4 | 0.00 | 0.036 |
| 614 | Cytophagaceae | 1.35 × 10−4 | 0.00 | 0.038 | 1.35 × 10−4 | 0.00 | 0.036 |
| 668 | Flavobacteriaceae | 1.22 × 10−4 | 0.00 | 0.046 | 1.22 × 10−4 | 0.00 | 0.046 |
| Surface | Subsurface | ||||
|---|---|---|---|---|---|
| OTU | Identity | p-value | OTU | Identity | p-value |
| 2 | Halobacteriaceae | 0.014 | 11 | Nitrososphaera | 0.009 |
| 4 | Halobacteriaceae | 0.023 | 42 | Nitrososphaera | 0.008 |
| 19 | Halobacteriaceae | 0.035 | 60 | Methanomassiliicoccaceae | 0.028 |
| 22 | Haloferacaceae | 0.033 | |||
| 23 | Halobacteriaceae | 0.030 | |||
| 25 | Halobacteriaceae | 0.022 | |||
| 27 | Haloferacaceae | 0.036 | |||
| 45 | Halobacteriaceae | 0.043 | |||
| Hydrated | |||||
| OTU | Identity | p-value | |||
| 1 | Halobacteriaceae | 0.023 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weingarten, E.A.; Lawson, L.A.; Jackson, C.R. The Saltpan Microbiome Is Structured by Sediment Depth and Minimally Influenced by Variable Hydration. Microorganisms 2020, 8, 538. https://doi.org/10.3390/microorganisms8040538
Weingarten EA, Lawson LA, Jackson CR. The Saltpan Microbiome Is Structured by Sediment Depth and Minimally Influenced by Variable Hydration. Microorganisms. 2020; 8(4):538. https://doi.org/10.3390/microorganisms8040538
Chicago/Turabian StyleWeingarten, Eric A., Lauren A. Lawson, and Colin R. Jackson. 2020. "The Saltpan Microbiome Is Structured by Sediment Depth and Minimally Influenced by Variable Hydration" Microorganisms 8, no. 4: 538. https://doi.org/10.3390/microorganisms8040538
APA StyleWeingarten, E. A., Lawson, L. A., & Jackson, C. R. (2020). The Saltpan Microbiome Is Structured by Sediment Depth and Minimally Influenced by Variable Hydration. Microorganisms, 8(4), 538. https://doi.org/10.3390/microorganisms8040538