Transcriptome Analysis of Gene Expression in Dermacoccus abyssi HZAU 226 under Lysozyme Stress


Abstract
:1. Introduction
2. Materials and Methods
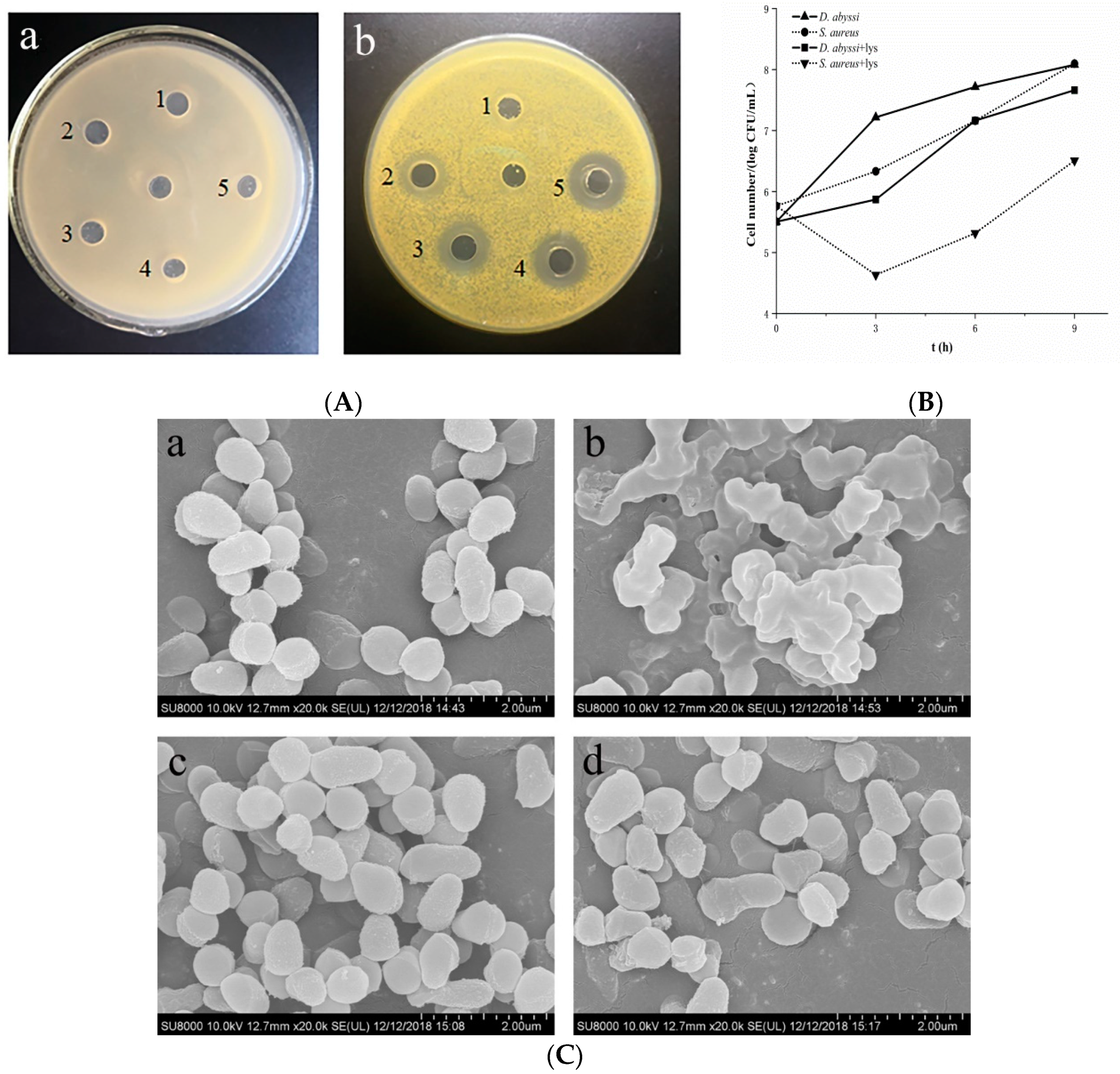
2.1. Characterization of Lysozyme Resistance
2.2. Lysozyme Treatment, RNA Extraction, Illumina Library Construction and Sequencing
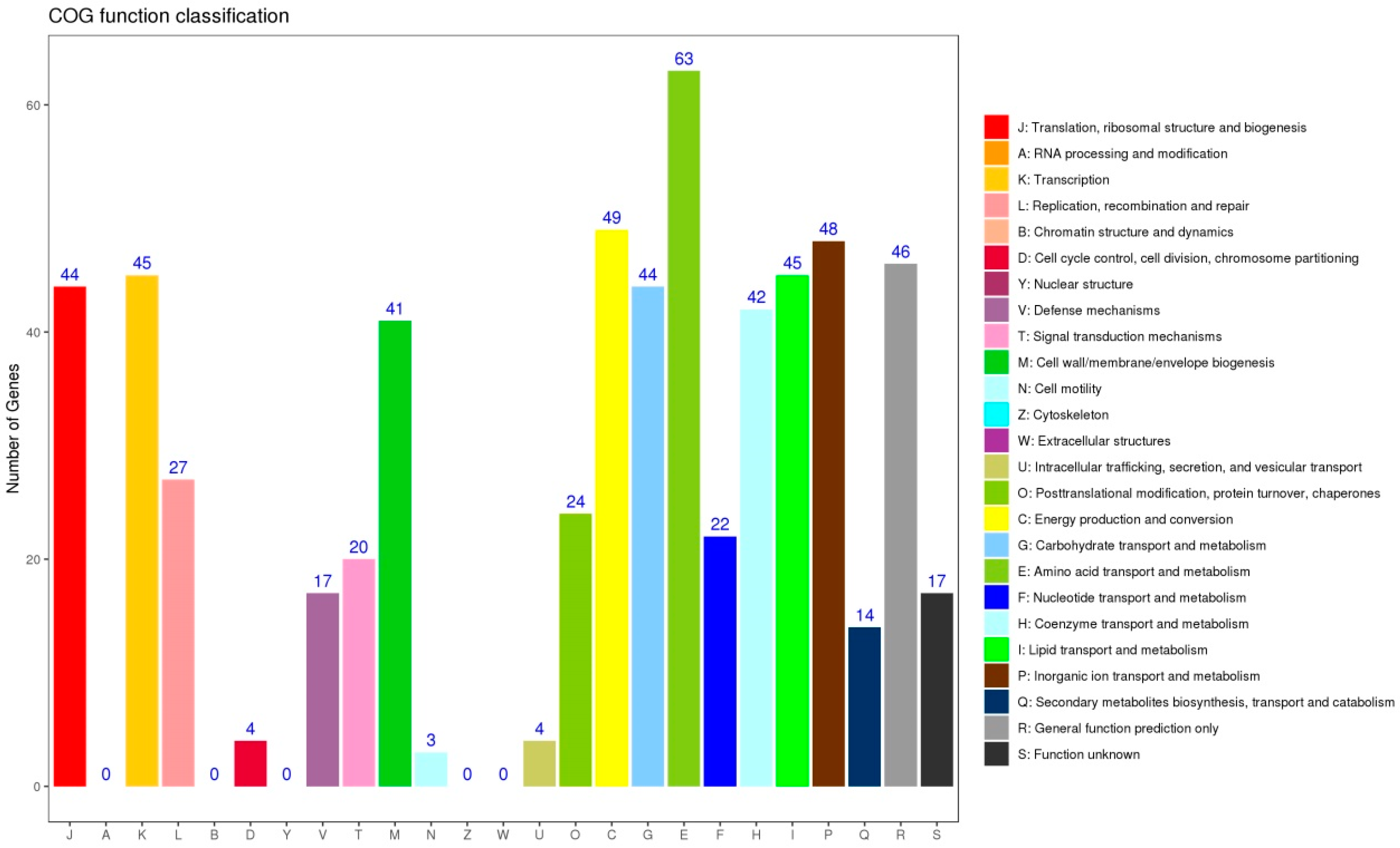
2.3. Sequence Analysis and Functional Annotation
3. Results
3.1. High Lysozyme Resistance of D. abyssi HZAU 226
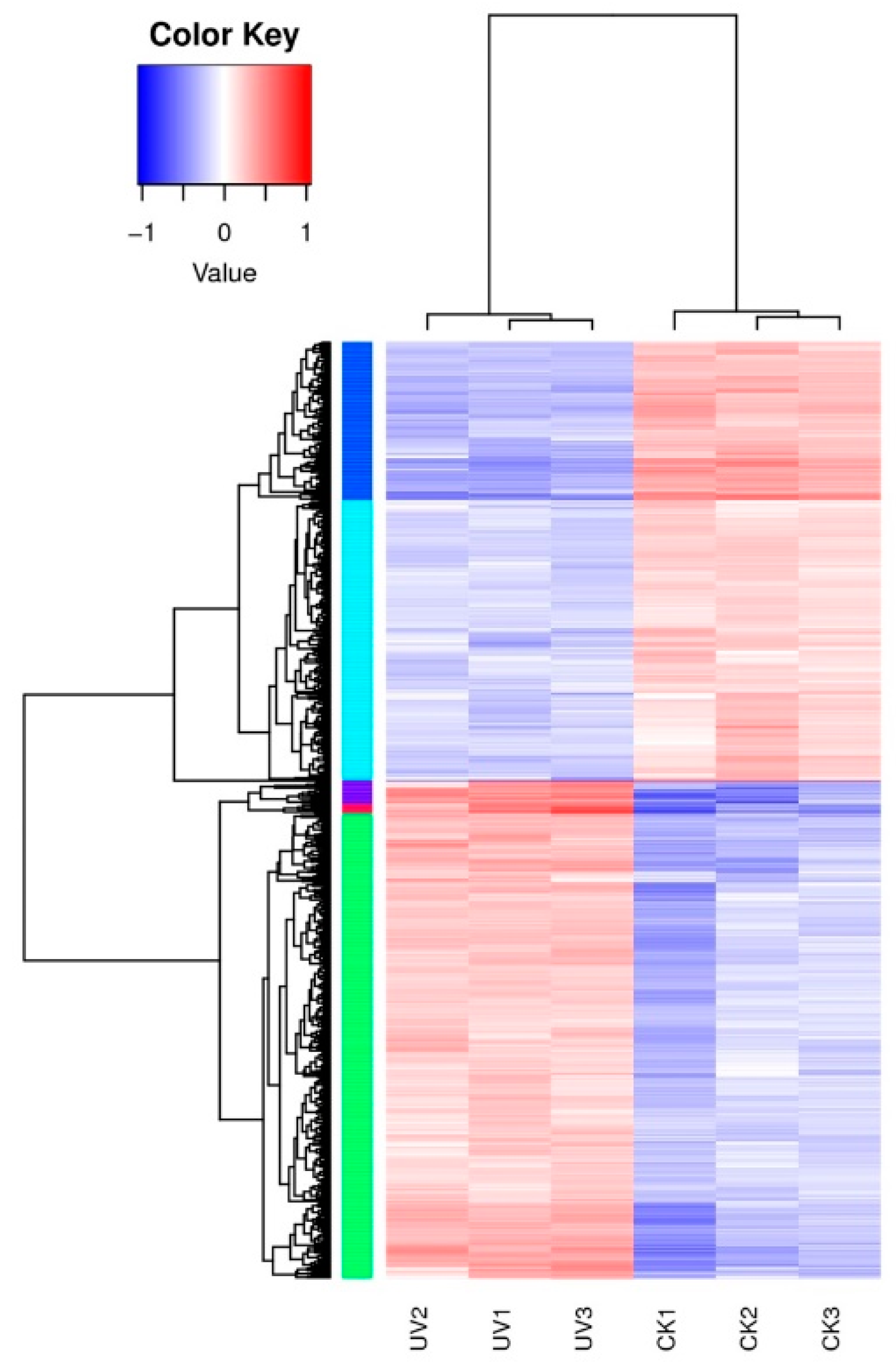
3.2. Quality Control of Sequencing Data
3.3. Identified New Genes and sRNA by Transcriptome Analysis
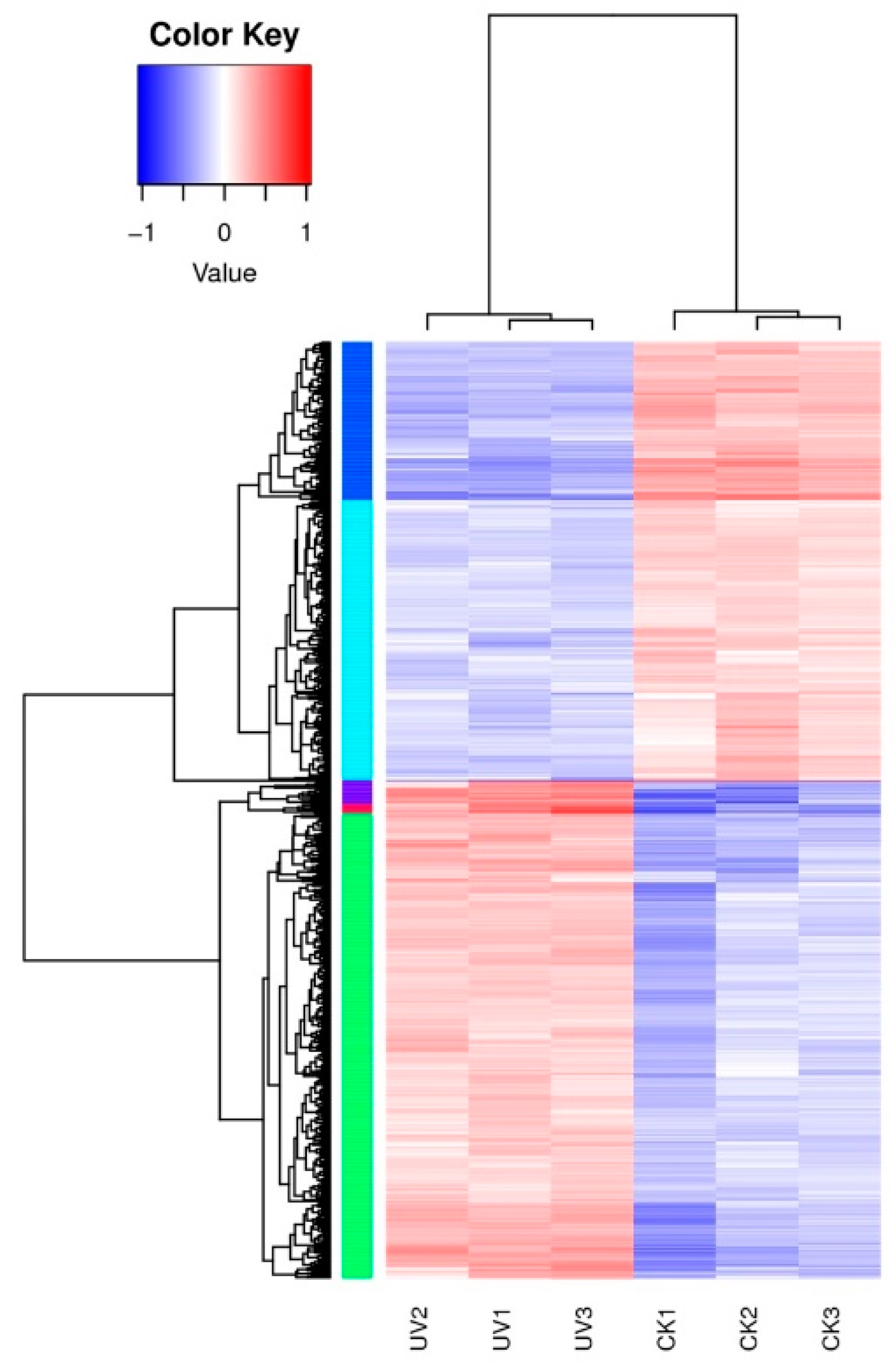
3.4. Differentially Expressed Genes’ (DEGs) Response to Lysozyme Treatment
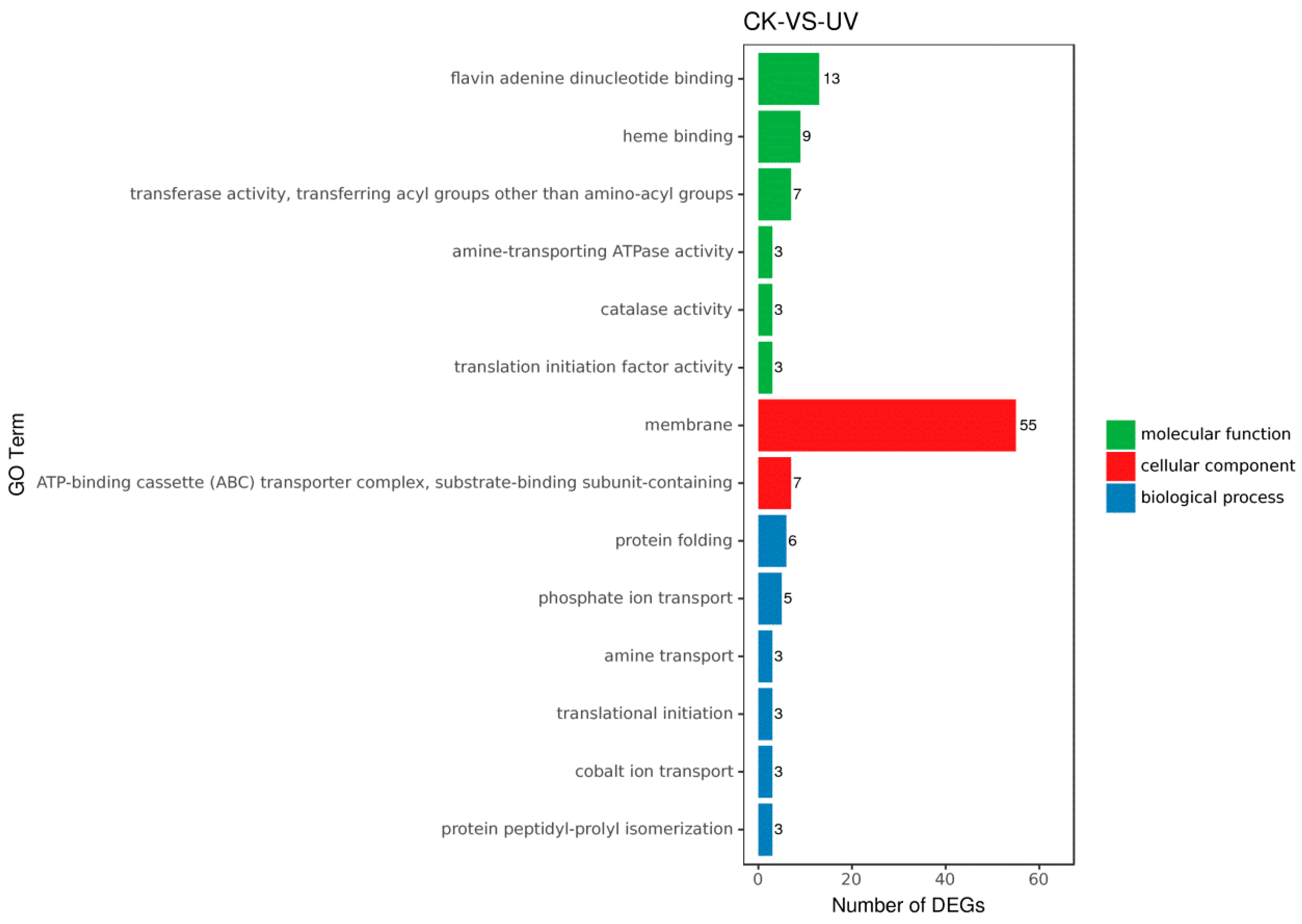
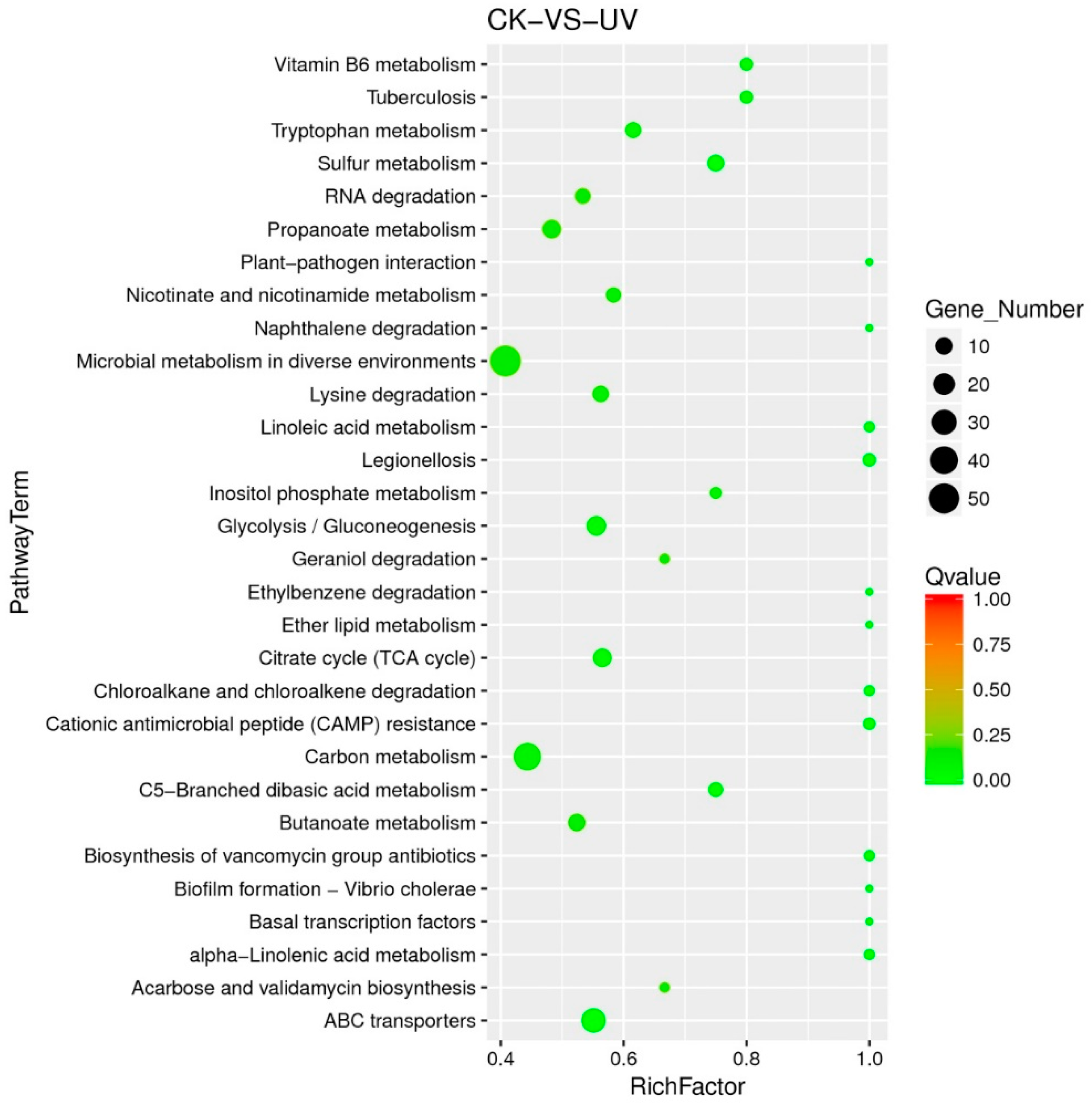
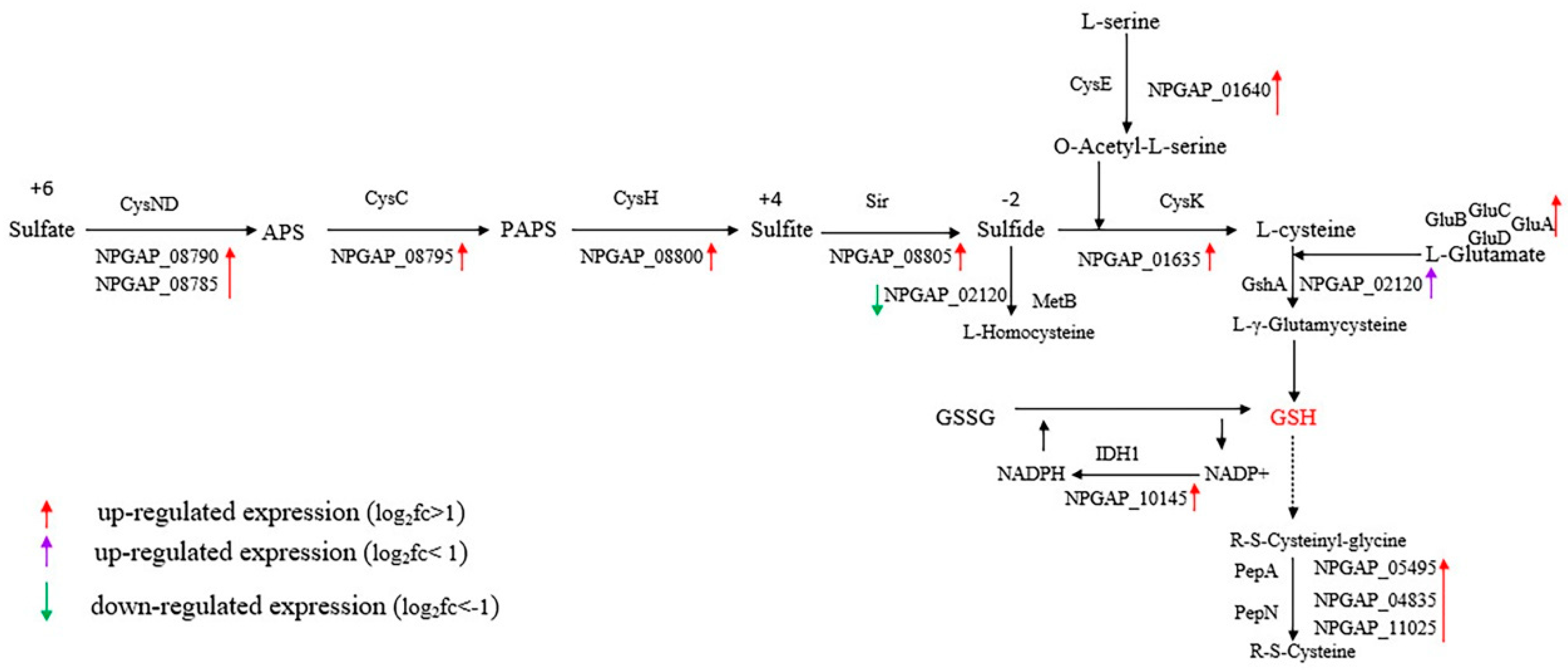
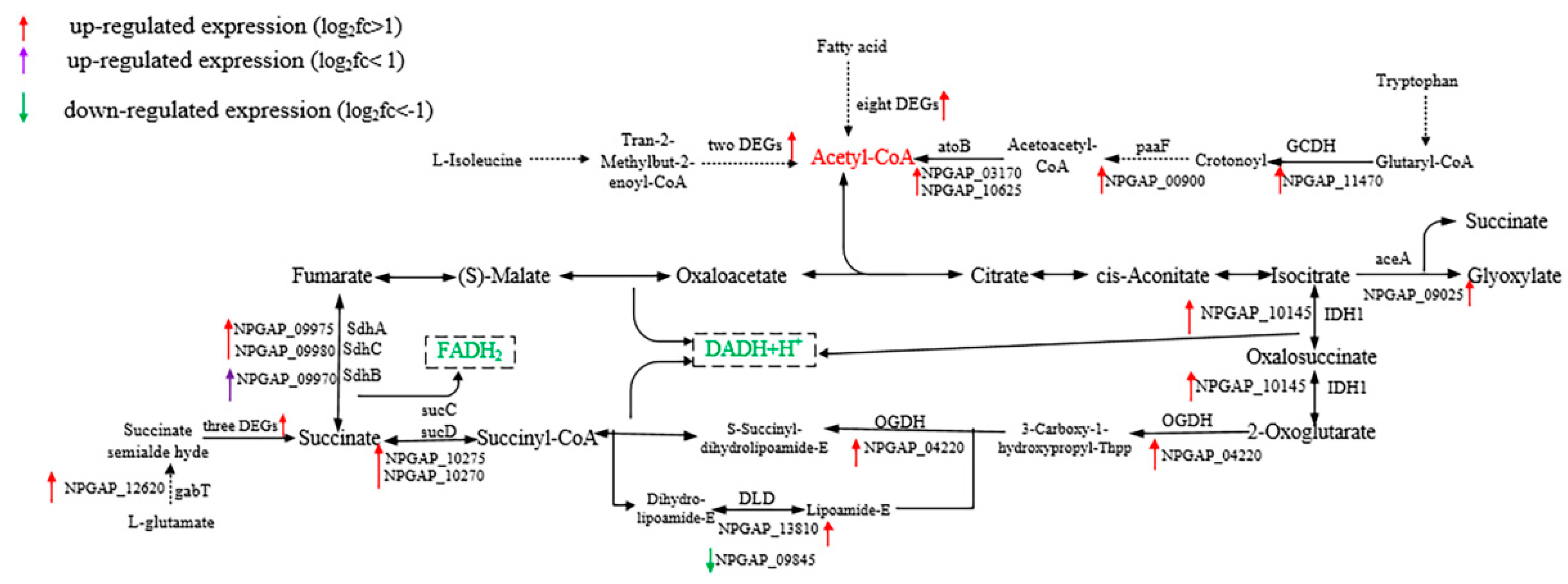
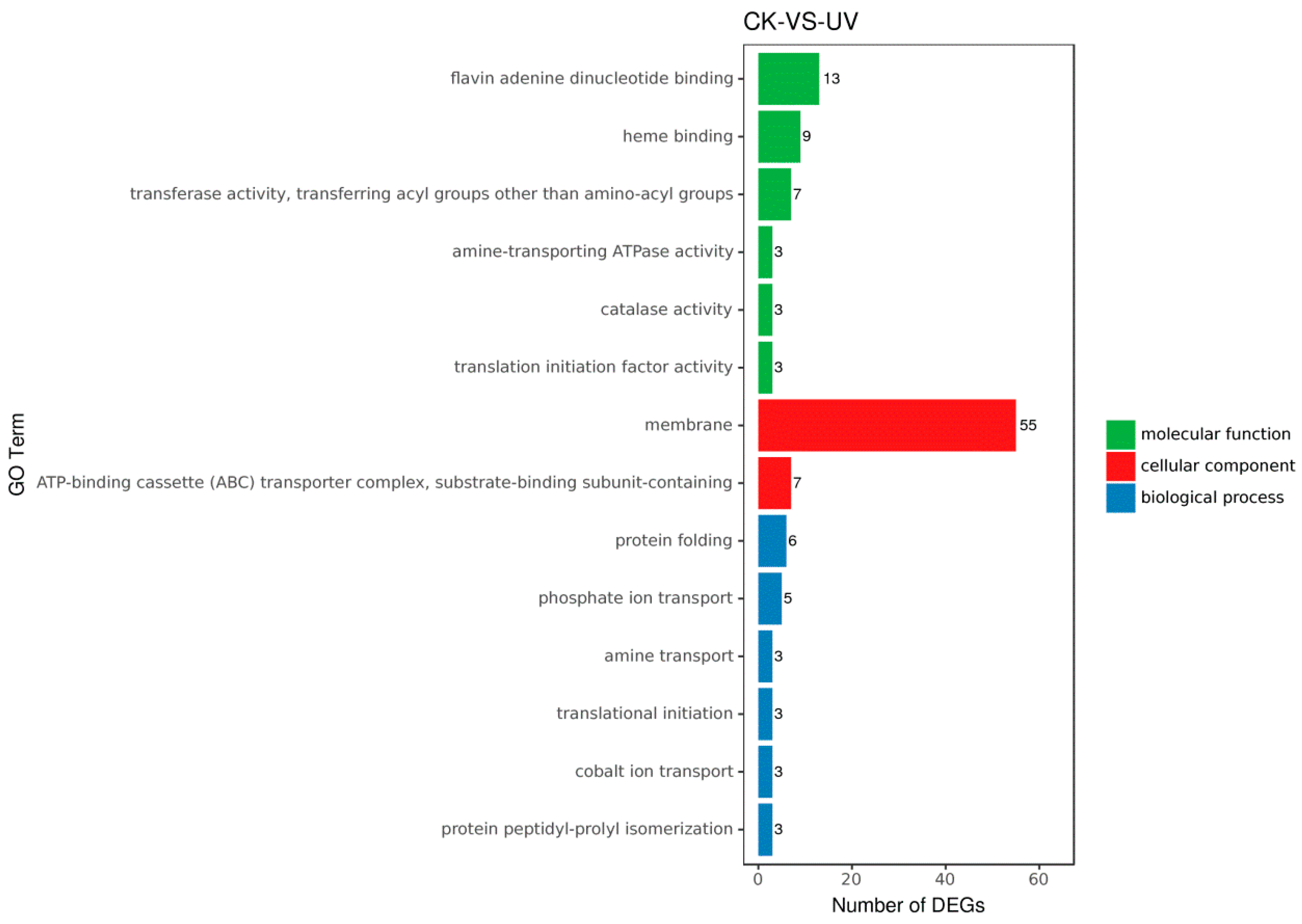
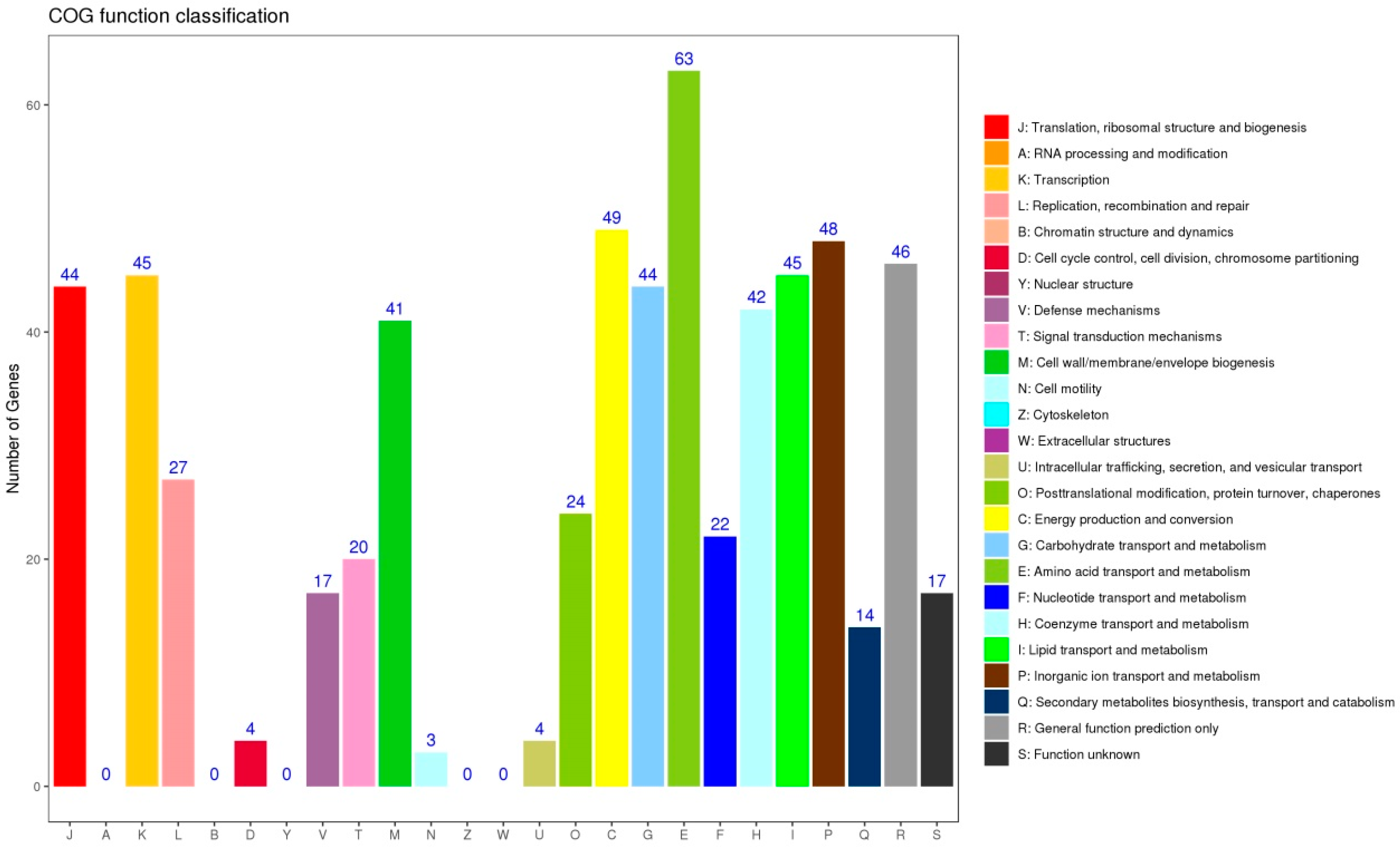
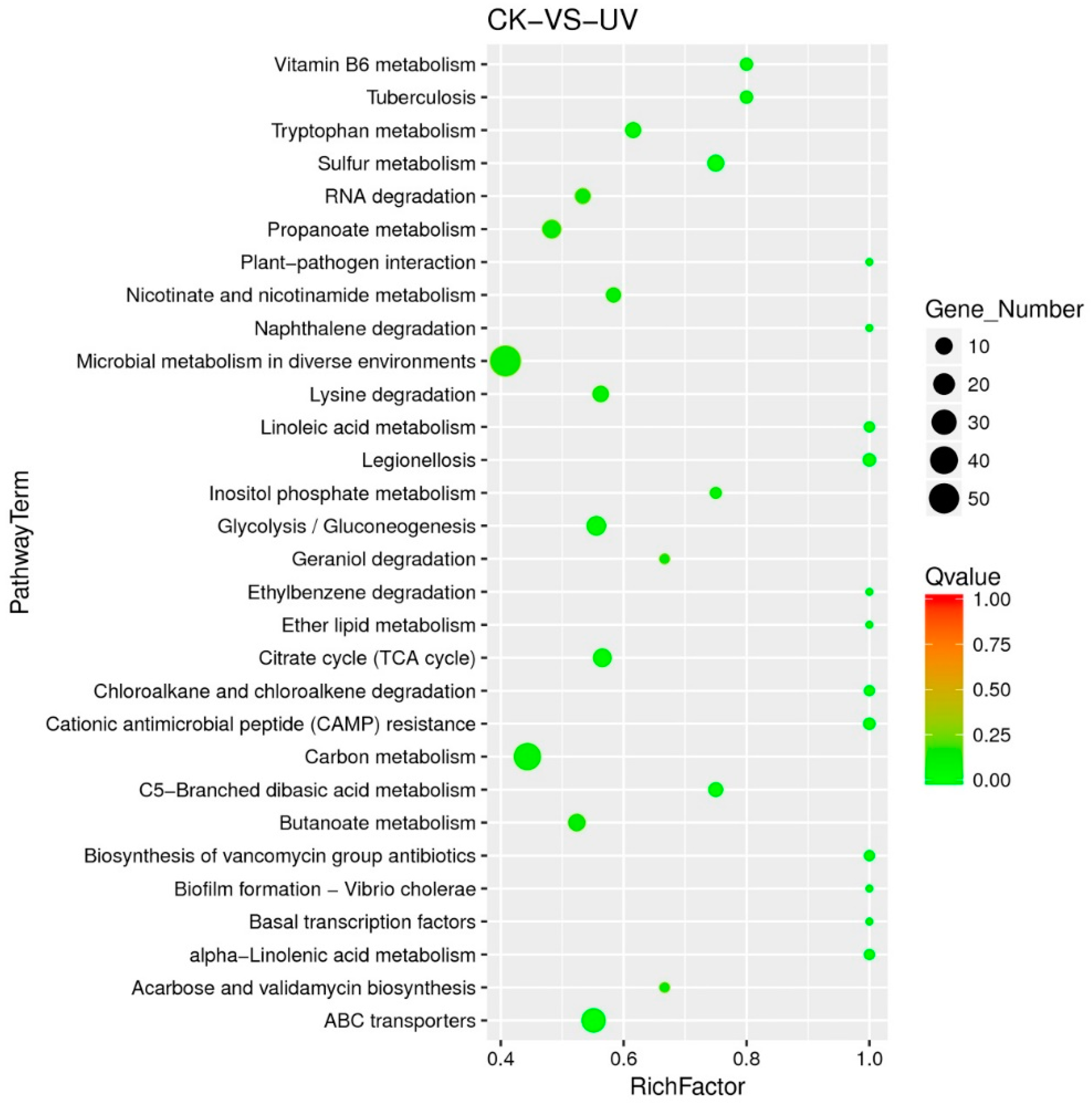
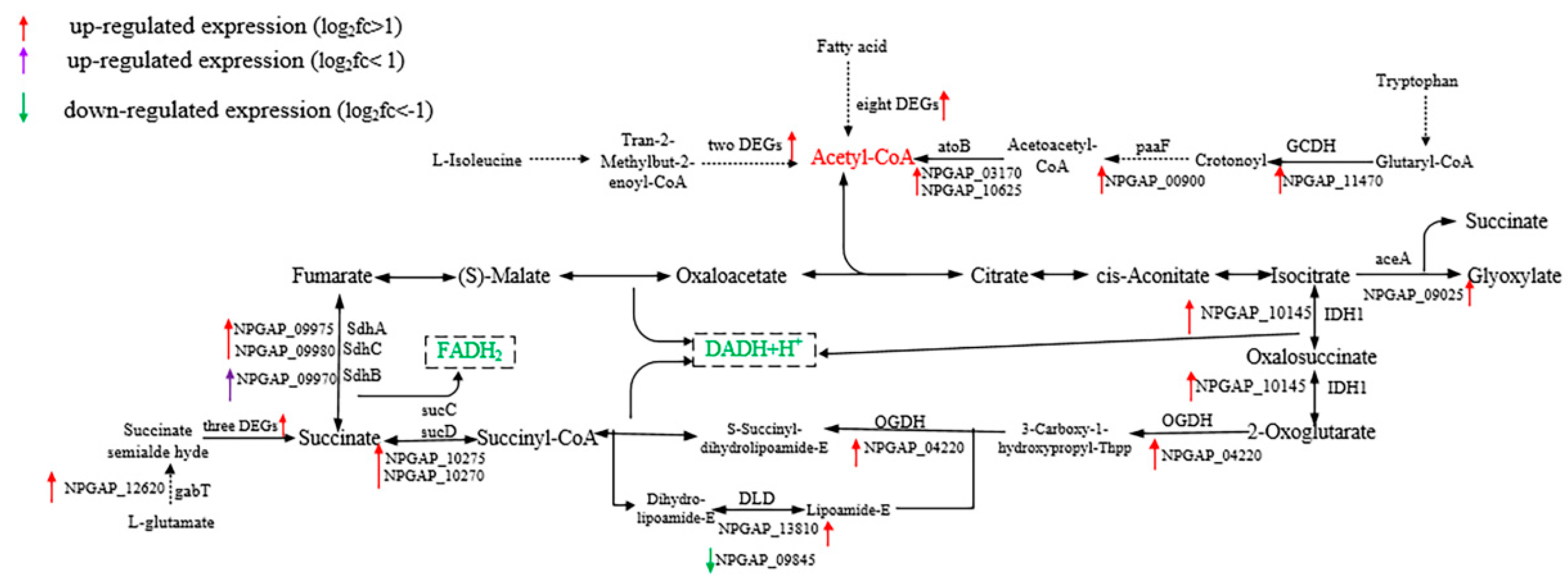
3.5. Functional Analysis of DEGs
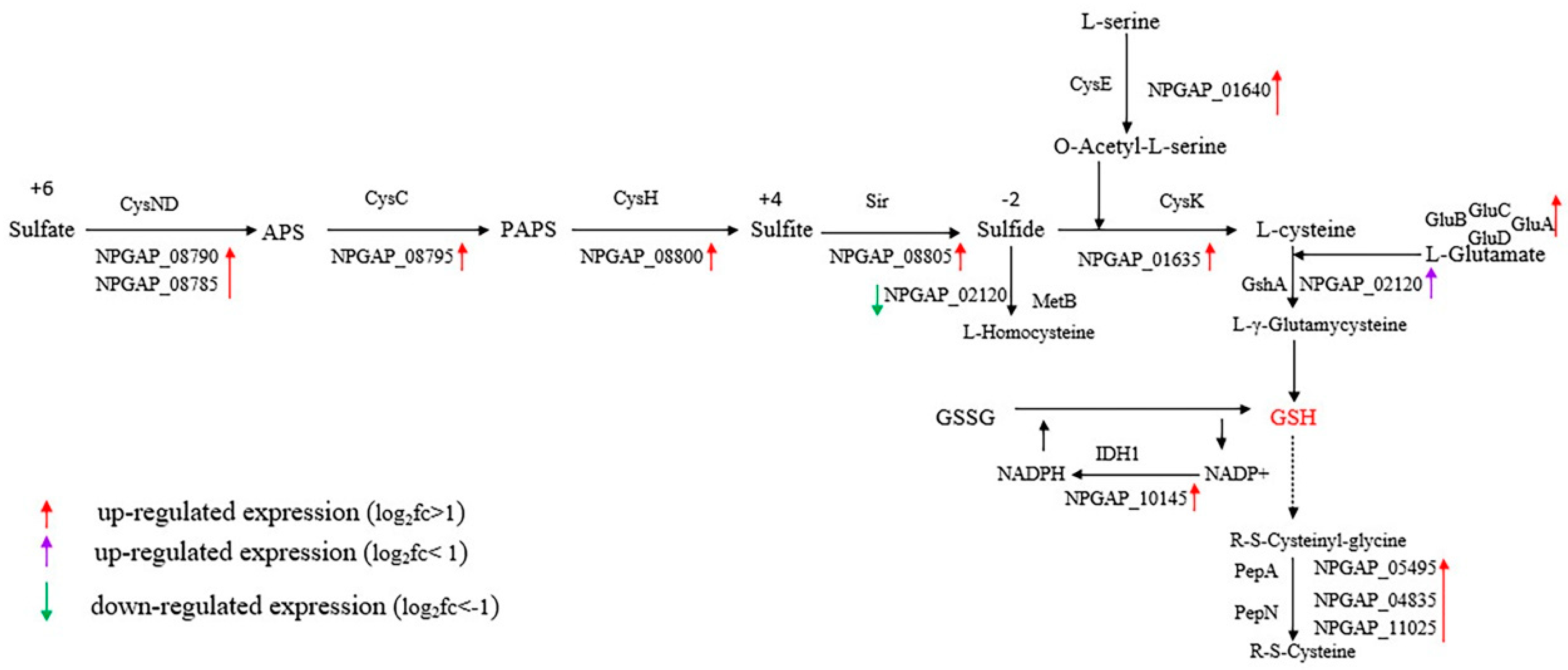
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Parte, A.C. LPSN—List of prokaryotic names with standing in nomenclature (bacterio.net), 20 years on. Int. J. Syst. Evol. Microbiol. 2018, 68, 1825–1829. [Google Scholar] [CrossRef]
- Pathom-aree, W.; Nogi, Y.; Sutcliffe, I.C.; Ward, A.C.; Horikoshi, K.; Bull, A.T.; Goodfellow, M. Williamsia marianensis sp. nov., a novel actinomycete isolated from the Mariana Trench. Int. J. Syst. Evol. Microbiol. 2006, 56, 1123–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Mageed, W.M.; Milne, B.F.; Wagner, M.; Schumacher, M.; Sandor, P.; Pathom-aree, W.; Goodfellow, M.; Bull, A.T.; Horikoshi, K.; Ebel, R.; et al. Dermacozines, a new phenazine family from deep-sea dermacocci isolated from a Mariana Trench sediment. Org. Biomol. Chem. 2010, 8, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Lang, W.; Sirisansaneeyakul, S.; Martins, L.O.; Ngiwsara, L.; Sakairi, N.; Pathom-aree, W.; Okuyama, M.; Mori, H.; Kimura, A. Biodecolorization of a food azo dye by the deep sea Dermacoccus abyssi MT1.1T strain from the Mariana Trench. J. Environ. Manag. 2014, 132, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Huang, Z.; Lei, Y.; Zhang, L.; Li, Y.; Luo, Y. Application of Illumina-MiSeq high throughput sequencing and culture-dependent techniques for the identification of microbiota of silver carp (hypophthalmichthys molitrix) treated by tea polyphenols. Food Microbiol. 2018, 76, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Q.; Gu, L.; Ruan, Y.; Zhang, X.; Guo, A. identification of a spoilage bacterium s from eggs with tolerance to egg white protein and de novo sequencing analysis of the strain. Mod. Food Sci. Technol. 2019, 35, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Vocadlo, D.J.; Davies, G.J.; Laine, R.; Withers, S.G.J.N. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar] [CrossRef]
- Herbert, S.; Bera, A.; Nerz, C.; Kraus, D.; Peschel, A.; Goerke, C.; Meehl, M.; Cheung, A.; Goetz, F. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog. 2007, 3, 981–994. [Google Scholar] [CrossRef]
- Kumaresan, V.; Bhatt, P.; Arockiaraj, J. Membrane disruption antimicrobial mechanism of Channa striatus lysozyme-derived antimicrobial peptides (AMP). Fish Shellfish Immunol. 2016, 53, 74–75. [Google Scholar] [CrossRef]
- Carrillo, W.; Ramos, M. Identification of antimicrobial peptides of native and heated hydrolysates from hen egg white lysozyme. J. Med. Food 2018, 21, 915–926. [Google Scholar] [CrossRef]
- Juneja, V.K.; Dwivedi, H.P.; Xianghe, Y. Novel natural food antimicrobials. Annu. Rev. Food Sci. Technol. 2012, 3, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.M.; Weiser, J.N. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect. Immun. 2011, 79, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmer, W. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 2008, 32, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Tomasz, A. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 2000, 275, 20496–20501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynihan, P.J.; Sychantha, D.; Clarke, A.J. Chemical biology of peptidoglycan acetylation and deacetylation. Bioorg. Chem. 2014, 54, 44–50. [Google Scholar] [CrossRef]
- Sychantha, D.; Jones, C.S.; Little, D.J.; Moynihan, P.J.; Robinson, H.; Galley, N.F.; Roper, D.I.; Dowson, C.G.; Howell, P.L.; Clarke, A.J. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Bera, A.; Biswas, R.; Herbert, S.; Kulauzovic, E.; Weidenmaier, C.; Peschel, A.; Goetz, F. Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J. Bacteriol. 2007, 189, 280–283. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, T.A.; Ludovice, A.M.; Sobral, R.G. Contribution of Peptidoglycan Amidation to Beta-Lactam and Lysozyme Resistance in Different Genetic Lineages of Staphylococcus aureus. Microb. Drug Resist. 2014, 20, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Monchois, V.; Abergel, C.; Sturgis, J.; Jeudy, S.; Claverie, J.M. Escherichia coli ykfE ORFan gene encodes a potent inhibitor of C-type lysozyme. J. Biol. Chem. 2001, 276, 18437–18441. [Google Scholar] [CrossRef] [Green Version]
- Callewaert, L.; Aertsen, A.; Deckers, D.; Vanoirbeek, K.G.A.; Vanderkelen, L.; Van Herreweghe, J.M.; Masschalck, B.; Nakimbugwe, D.; Robben, J.; Michiels, C.W. A new family of lysozyme inhibitors contributing to lysozyme tolerance in gram-negative bacteria. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielke, R.A.; Le Van, A.; Baarda, B.I.; Herrera, M.F.; Acosta, C.J.; Jerse, A.E.; Sikora, A.E. SliC is a surface-displayed lipoprotein that is required for the anti-lysozyme strategy during Neisseria gonorrhoeae infection. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Li, W.; Shi, G.; Li, H.; Yang, X.; Xia, Z.; Ren, Y.; Li, Z.; Chen, H.; Bei, W. The VraSR regulatory system contributes to virulence in Streptococcus suis via resistance to innate immune defenses. Virulence 2018, 9, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogos, A.; Cristobal Jimenez, J.; Chang, J.C.; Wilkening, R.V.; Federle, M.J. A quorum sensing-regulated protein binds cell wall components and enhances lysozyme resistance in Streptococcus pyogenes. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [Green Version]
- Benachour, A.; Ladjouzi, R.; Le Jeune, A.; Hebert, L.; Thorpe, S.; Courtin, P.; Chapot-Chartier, M.-P.; Prajsnar, T.K.; Foster, S.J.; Mesnage, S. The lysozyme-induced peptidoglycan n-acetylglucosamine deacetylase pgda (EF1843) is required for enterococcus faecalis virulence. J. Bacteriol. 2012, 194, 6066–6073. [Google Scholar] [CrossRef] [Green Version]
- Nhat Khai, B.; Turk, S.; Buckenmaier, S.; Stevenson-Jones, F.; Zeuch, B.; Gobec, S.; Vollmer, W. Development of screening assays and discovery of initial inhibitors of pneumococcal peptidoglycan deacetylase PgdA. Biochem. Pharmacol. 2011, 82, 43–52. [Google Scholar]
- Bhattacharjee, N.; Feliks, M.; Shaik, M.M.; Field, M.J. Catalytic mechanism of peptidoglycan deacetylase: A computational study. J. Phys. Chem. B 2017, 121, 89–99. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Yujuan, S.; Shigang, G.; Baranzoni, G.M.; Yanping, X.; Yanhong, L. Comparative transcriptome RNA-Seq analysis of Listeria monocytogenes with sodium lactate adaptation. Food Control 2018, 91, 193–201. [Google Scholar]
- Hagi, T.; Kobayashi, M.; Nomura, M. Whole-transcriptome analysis of oxidative stress response genes in carotenoid-producing enterococcus gilvus. Biosci. Biotechnol. Biochem. 2018, 82, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Li, W.; Ying, X.; Lu, Q.; Chen, L. Predicting sRNAs and their targets in bacteria. Genomics Proteomics Bioinform. 2012, 10, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Varet, H.; Brillet-Gueguen, L.; Coppee, J.-Y.; Dillies, M.-A. SARTools: A DESeq2-and edgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Woodhall, M.R.; Alvarez, J.; Cartron, M.L.; Andrews, S.C. EfeUOB (YcdNOB) is a tripartite, acid-induced and CpxAR-regulated, low-pH Fe2+ transporter that is cryptic in Escherichia coli K-12 but functional in E-coli O157: H7. Mol. Microbiol. 2007, 65, 857–875. [Google Scholar] [CrossRef]
- Turlin, E.; Debarbouille, M.; Augustyniak, K.; Gilles, A.-M.; Wandersman, C. Staphylococcus aureus FepA and FepB proteins drive heme iron utilization in Escherichia coli. PLoS ONE 2013, 8, e56529. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, M.; Cheng, A. bacterial iron uptake system and anti-infectious immunity of the host. Life Sci. Res. 2016, 20, 82–88. [Google Scholar]
- Labudda, M.; Azam, F.M.S. Glutathione-dependent responses of plants to drought: A review. Acta Soc. Bot. Pol. 2014, 83, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.-Y.; Chen, J.; Li, Y. The function of glutathione/glutathione peroxidase system in the oxidative stress resistance systems of microbial cells. Chin. J. Biotechnol. 2007, 23, 770–775. [Google Scholar] [CrossRef]
- Yang, J.; Kim, M.J.; Yoon, W.; Kim, E.Y.; Kim, H.; Lee, Y.; Min, B.; Kang, K.S.; Son, J.H.; Park, H.T.; et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+- dependent isocitrate dehydrogenase (IDH). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef]
- Konings, W.N. The cell membrane and the struggle for life of lactic acid bacteria. In Lactic Acid Bacteria: Genetics, Metabolism and Applications; Springer: Dordrecht, Holland, 2002; pp. 3–27. [Google Scholar]
- Smits, S.H.J.; Hoeing, M.; Lecher, J.; Jebbar, M.; Schmitt, L.; Bremer, E. The compatible-solute-binding protein OpuAC from Bacillus subtilis: Ligand binding, site-directed mutagenesis, and crystallographic studies. J. Bacteriol. 2008, 190, 5663–5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wang, L.; Xie, M. Regulating inorganic phosphate transport and mediating bacterial resistance by PstS and PstB. Weishengwu Xuebao 2019, 59, 1429–1436. (In Chinese) [Google Scholar]
- Yan, X.; Budin-Verneuil, A.; Verneuil, N.; Gilmore, M.S.; Artigaud, S.; Auffray, Y.; Pichereau, V. Transcriptomic response of Enterococcus faecalis V583 to low hydrogen peroxide levels. Curr. Microbiol. 2015, 70, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-S.; Bi, Y.-T.; Dong, C.; Yang, J.-F.; Liang, W.-D. Transcriptome Analysis of adaptive heat shock response of Streptococcus thermophilus. PLoS ONE 2011, 6, e25777. [Google Scholar] [CrossRef] [PubMed]
- Grimm, I.; Dumke, J.; Dreier, J.; Knabbe, C.; Vollmer, T. Biofilm formation and transcriptome analysis of Streptococcus gallolyticus subsp gallolyticus in response to lysozyme. PLoS ONE 2018, 13, e0191705. [Google Scholar] [CrossRef] [Green Version]
- Wall, D.M.; Duffy, P.S.; DuPont, C.; Prescott, J.F.; Meijer, W.G. Isocitrate lyase activity is required for virulence of the intracellular pathogen Rhodococcus Equi. Infect. Immun. 2005, 73, 6736–6741. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.-M.; Lee, H.-I.; Sadowsky, M.J.; Sugawara, M.; Chang, W.-S. Characterization of a functional role of the bradyrhizobium japonicum isocitrate lyase in desiccation tolerance. Int. J. Mol. Sci. 2015, 16, 16695–16709. [Google Scholar] [CrossRef] [Green Version]
- Bai, Z.-Y.; Wang, T.; Wu, Y.-H.; Wang, K.; Liang, Q.-Y.; Pan, Y.-Z.; Jiang, B.-B.; Zhang, L.; Liu, G.-L.; Jia, Y.; et al. Whole-transcriptome sequence analysis of differentially expressed genes in Phormium tenax under drought stress. Sci. Rep. 2017, 7, 1–9. [Google Scholar]
- Wang, J.; Zhu, J.; Zhang, Y.; Fan, F.; Li, W.; Wang, F.; Zhong, W.; Wang, C.; Yang, J. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of indica rice at seedling stage. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rismondo, J.; Wamp, S.; Aldridge, C.; Vollmer, W.; Halbedel, S. Stimulation of PgdA-dependent peptidoglycan N-deacetylation by GpsB-PBP A1 in Listeria monocytogenes. Mol. Microbiol. 2018, 107, 472–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morlot, C.; Straume, D.; Peters, K.; Hegnar, O.A.; Simon, N.; Villard, A.-M.; Contreras-Martel, C.; Leisico, F.; Breukink, E.; Gravier-Pelletier, C.; et al. Structure of the essential peptidoglycan amidotransferase MurT/GatD complex from Streptococcus pneumoniae. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muench, D.; Roemer, T.; Lee, S.H.; Engeser, M.; Sahl, H.G.; Schneider, T. Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PLoS Pathog. 2012, 8, e1002509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.S.; Chowdhury, C.; Nelson, D.E. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 2008, 16, 309–317. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sample Description | Total Reads | Bases | Q20 1 (%) | Q30 2 (%) | GC 3 (%) | Total Mapped |
|---|---|---|---|---|---|---|---|
| CK1 | Control replication 1 | 33,366,114 | 4,934,245,447 | 96.80 | 91.36 | 67.43 | 31,528,403 (94.4923%) |
| CK2 | Control replication 2 | 33,483,220 | 4,948,473,732 | 96.61 | 90.91 | 67.29 | 31,529,020 (94.1636%) |
| CK3 | Control replication 3 | 34,522,436 | 5,128,803,026 | 97.78 | 93.92 | 67.40 | 32,692,711 (94.6999%) |
| UV1 | Lysozyme treatment replication 1 | 56,645,366 | 8,421,131,651 | 98.17 | 94.79 | 66.31 | 51,680,501 (91.2352%) |
| UV2 | Lysozyme treatment replication 2 | 45,945,296 | 6,769,856,909 | 96.53 | 90.80 | 64.46 | 39,814,219 (86.6557%) |
| UV3 | Lysozyme treatment replication 3 | 38,707,694 | 5,712,404,289 | 96.60 | 90.93 | 64.77 | 33,415,978 (86.3290%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Ruan, Y.; Liu, W.; Chen, Q.; Gu, L.; Guo, A. Transcriptome Analysis of Gene Expression in Dermacoccus abyssi HZAU 226 under Lysozyme Stress. Microorganisms 2020, 8, 707. https://doi.org/10.3390/microorganisms8050707
Zhang X, Ruan Y, Liu W, Chen Q, Gu L, Guo A. Transcriptome Analysis of Gene Expression in Dermacoccus abyssi HZAU 226 under Lysozyme Stress. Microorganisms. 2020; 8(5):707. https://doi.org/10.3390/microorganisms8050707
Chicago/Turabian StyleZhang, Xinshuai, Yao Ruan, Wukang Liu, Qian Chen, Lihong Gu, and Ailing Guo. 2020. "Transcriptome Analysis of Gene Expression in Dermacoccus abyssi HZAU 226 under Lysozyme Stress" Microorganisms 8, no. 5: 707. https://doi.org/10.3390/microorganisms8050707
APA StyleZhang, X., Ruan, Y., Liu, W., Chen, Q., Gu, L., & Guo, A. (2020). Transcriptome Analysis of Gene Expression in Dermacoccus abyssi HZAU 226 under Lysozyme Stress. Microorganisms, 8(5), 707. https://doi.org/10.3390/microorganisms8050707