Microbiome of the Queensland Fruit Fly through Metamorphosis



Abstract
:1. Introduction
2. Results
2.1. Identification of Qfly
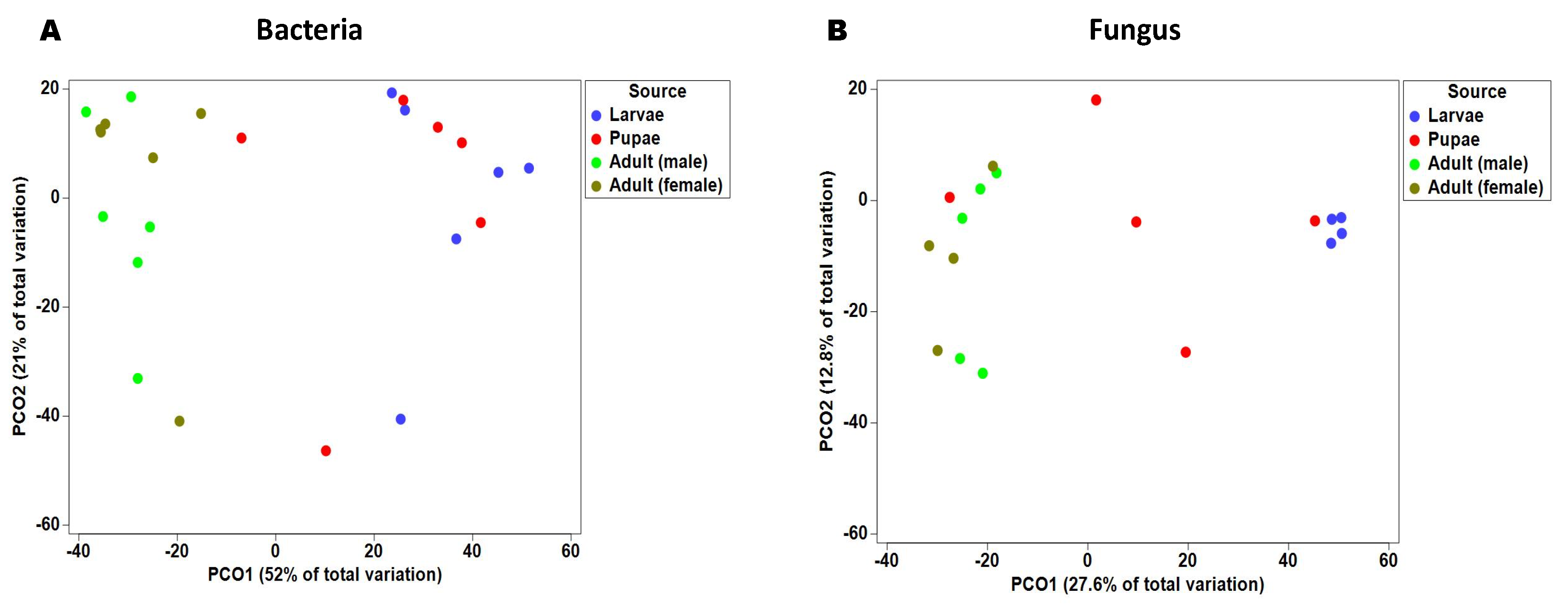
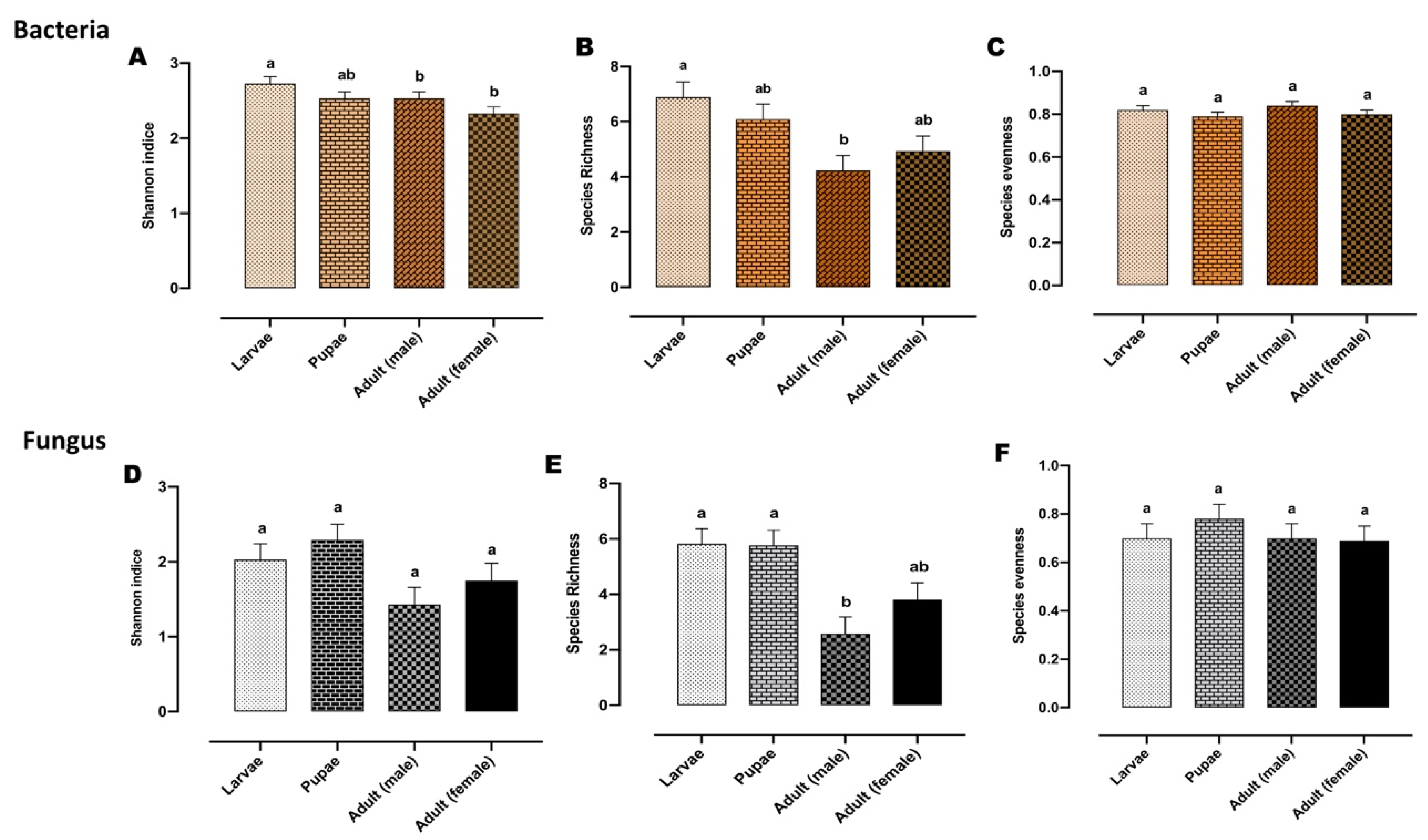
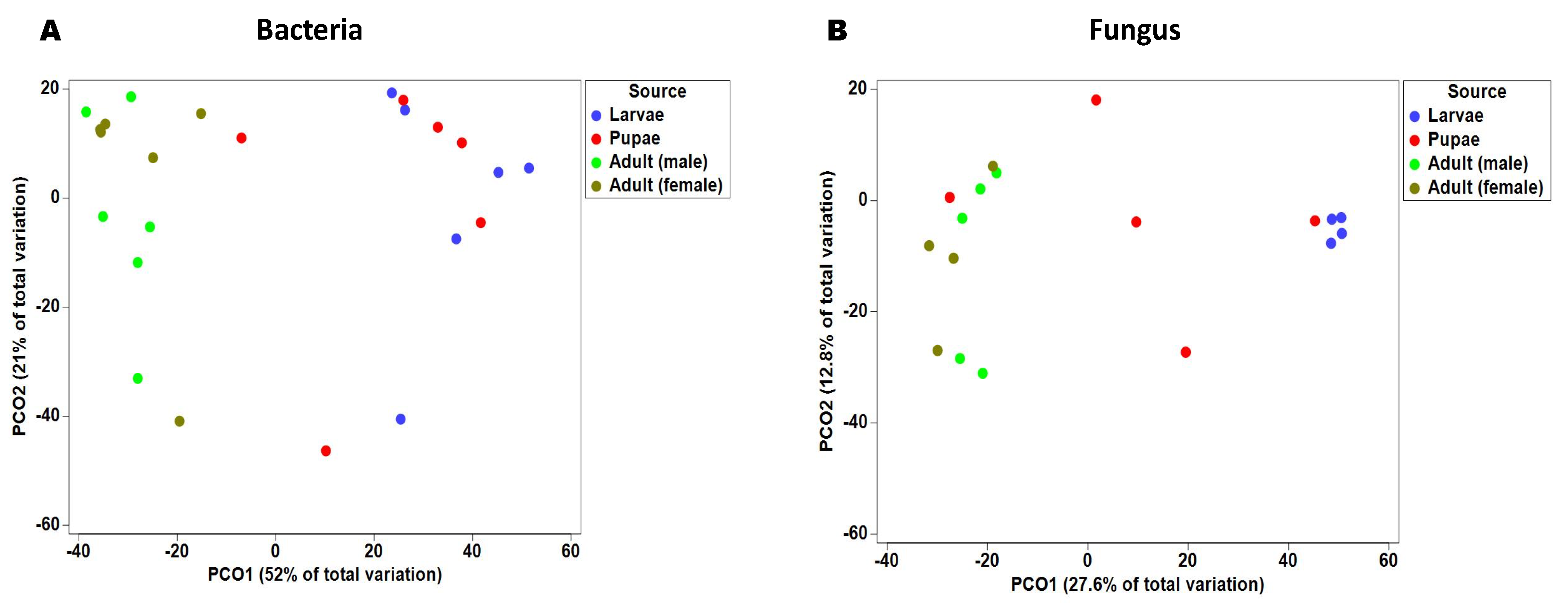
2.2. Gut Bacterial Alpha and Beta Diversity During Metamorphosis
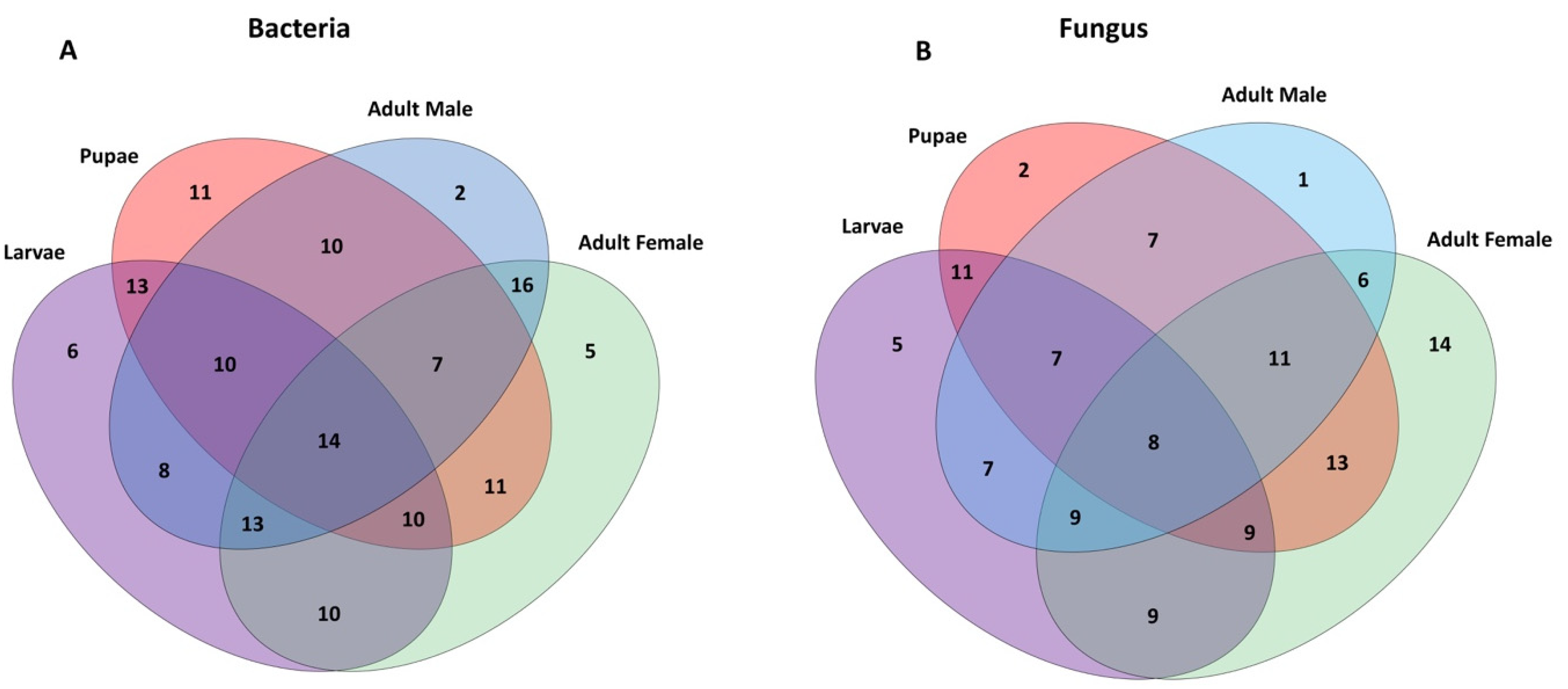
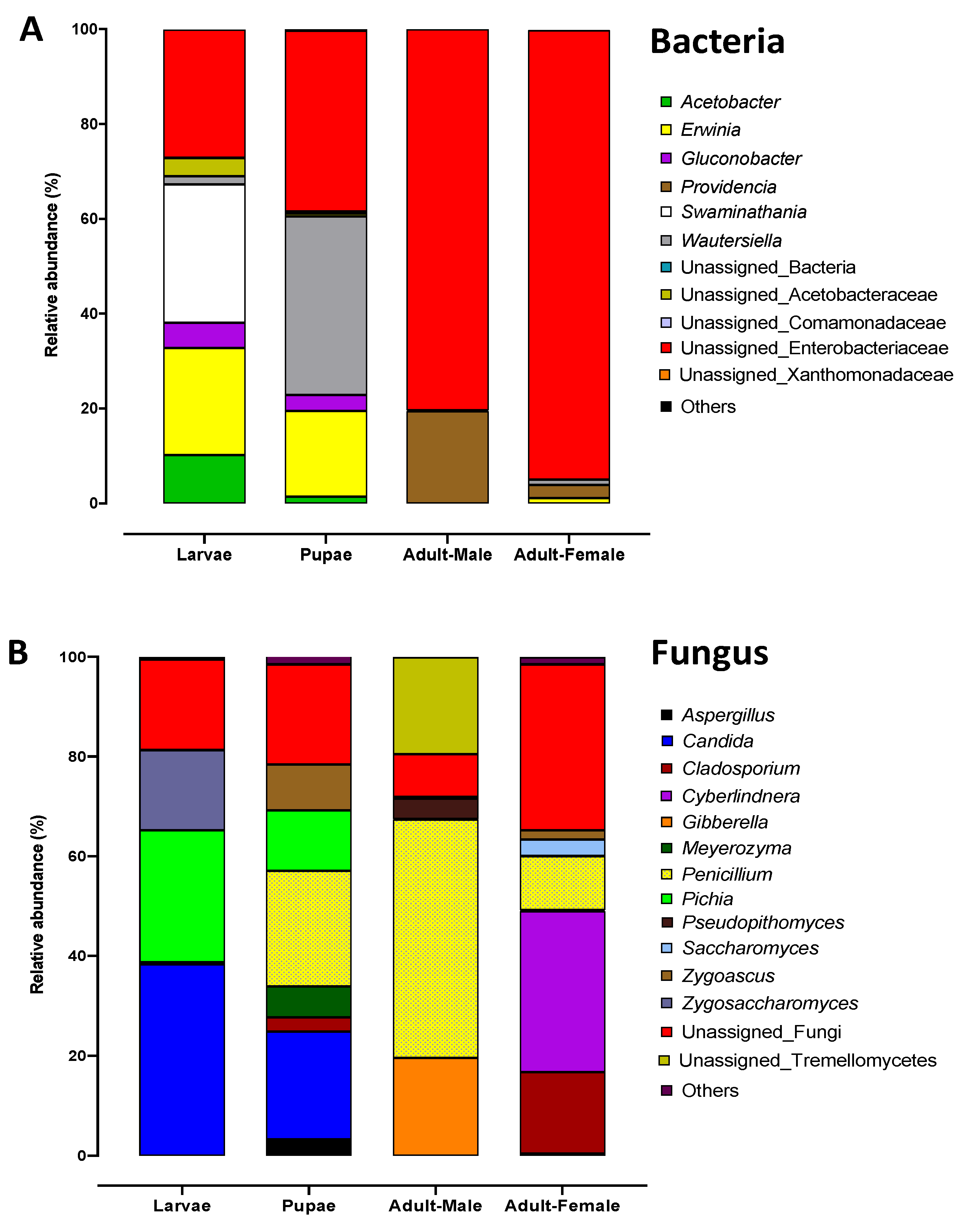
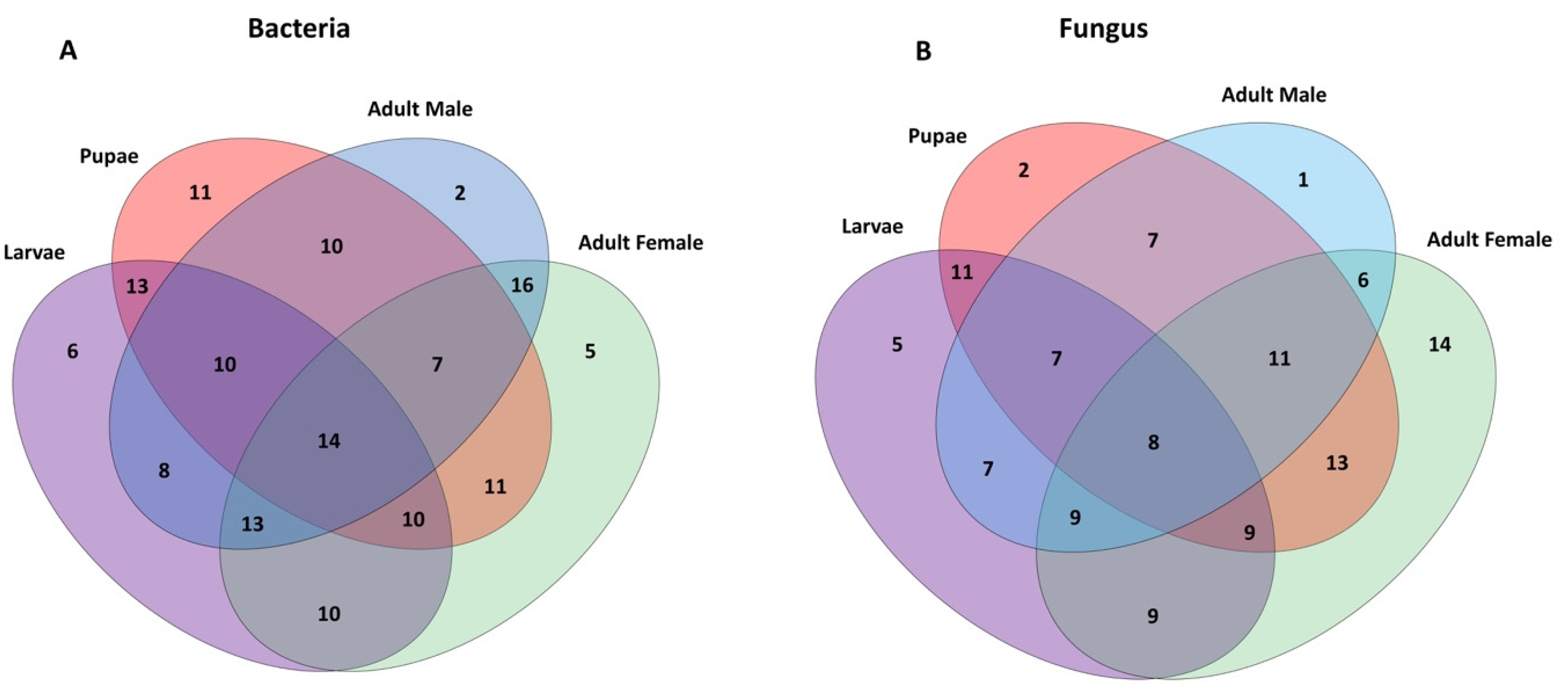
2.3. Bacterial Communities Associated with Metamorphosis
2.4. Fungal Alpha and Beta Diversity during Metamorphosis
2.5. Fungal Communities Associated with the Qfly Metamorphosis
3. Discussion
4. Materials and Methods
4.1. Qfly Sample Collection
4.2. Sample Preparation
4.3. Qfly Identification Using Mitochondrial Cytochrome Oxidase I (COI) Gene
4.4. Qfly Microbiome Profiling
4.5. Sequence Data Processing
4.6. Statistical Analysis
4.7. Data Availability Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Barnabas, B.; Blakesley, R.; Bouffard, G.; Brooks, S.; Coleman, H.; Dekhtyar, M. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottman, N.; Smidt, H.; De Vos, W.M.; Belzer, C. The function of our microbiota: Who is out there and what do they do? Front. Cell. Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [Green Version]
- Janson, E.M.; Stireman III, J.O.; Singer, M.S.; Abbot, P. Phytophagous insect–microbe mutualisms and adaptive evolutionary diversification. Evol. Int. J. Org. Evol. 2008, 62, 997–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.C.; Kim, S.H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.A.; Yoon, J.H.; Ryu, J.H.; Lee, W.J. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, R.; Masuya, H.; Endoh, R.; Kikuchi, T.; Ikeda, H. Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J. 2019, 13, 676. [Google Scholar] [CrossRef]
- Coon, K.L.; Vogel, K.J.; Brown, M.R.; Strand, M.R. Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 2014, 23, 2727–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenscraft, A.; Berry, M.; Hammer, T.; Peay, K.; Boggs, C. Structure and function of the bacterial and fungal gut microbiota of Neotropical butterflies. Ecol. Monogr. 2019, 89, e01346. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, W.; Zhu, F.; Wang, X.; Wang, X.; Lei, C. The gut microbiota in larvae of the housefly Musca domestica and their horizontal transfer through feeding. AMB Express 2017, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Tagliavia, M.; Messina, E.; Manachini, B.; Cappello, S.; Quatrini, P. The gut microbiota of larvae of Rhynchophorus ferrugineus Oliver (Coleoptera: Curculionidae). BMC Microbiol. 2014, 14, 136. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Cortés, N.; Quesada, M.; Watanabe, H.; Cano-Camacho, H.; Oyama, K. Endogenous plant cell wall digestion: A key mechanism in insect evolution. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 45–71. [Google Scholar] [CrossRef]
- Dillon, R.; Dillon, V. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Hammer, T.J.; Bowers, M.D. Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia 2015, 179, 1–14. [Google Scholar] [CrossRef]
- Chen, B.; Teh, B.S.; Sun, C.; Hu, S.; Lu, X.; Boland, W.; Shao, Y. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef]
- Galac, M.R.; Lazzaro, B.P. Comparative pathology of bacteria in the genus Providencia to a natural host, Drosophila melanogaster. Microbes Infect. 2011, 13, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.L.; Wang, J.B.; Brown, M.A.; Euerle, C.; Leger, R.J.S. Identification of Drosophila mutants affecting defense to an entomopathogenic fungus. Sci. Rep. 2015, 5, 12350. [Google Scholar] [CrossRef] [Green Version]
- Babendreier, D.; Joller, D.; Romeis, J.; Bigler, F.; Widmer, F. Bacterial community structures in honeybee intestines and their response to two insecticidal proteins. FEMS Microbiol. Ecol. 2007, 59, 600–610. [Google Scholar] [CrossRef]
- Mohr, K.I.; Tebbe, C.C. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ. Microbiol. 2006, 8, 258–272. [Google Scholar] [CrossRef]
- Ashbolt, N.J.; Inkerman, P.A. Acetic acid bacterial biota of the pink sugar cane mealybug, Saccharococcus sacchari, and its environs. Appl. Environ. Microbiol. 1990, 56, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Dowd, P. Symbiont-mediated detoxification in insect herbivores. Microb. Mediat. Plant Herbiv. Interact. 1991, 411, 440. [Google Scholar]
- Dowd, P.F. In situ production of hydrolytic detoxifying enzymes by symbiotic yeasts in the cigarette beetle (Coleoptera: Anobiidae). J. Econ. Entomol. 1989, 82, 396–400. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Suh, S.O.; Blackwell, M. Five novel Candida species in insect-associated yeast clades isolated from Neuroptera and other insects. Mycologia 2007, 99, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, I. Yeast-insect associations: It takes guts. Yeast 2018, 35, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, F.E.; Blackwell, M. Insect-Fungal Associations: Ecology and Evolution; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Grimaldi, D.; Engel, M.S.; Engel, M.S. Evolution of the Insects; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Johnston, P.R.; Rolff, J. Host and symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog. 2015, 11, e1005246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truman, J.W.; Riddiford, L.M. The origins of insect metamorphosis. Nature 1999, 401, 447. [Google Scholar] [CrossRef]
- Ventura, C.; Briones-Roblero, C.I.; Hernández, E.; Rivera-Orduña, F.N.; Zúñiga, G. Comparative analysis of the gut bacterial community of four Anastrepha fruit flies (Diptera: Tephritidae) based on pyrosequencing. Curr. Microbiol. 2018, 75, 966–976. [Google Scholar] [CrossRef]
- Yong, H.S.; Song, S.L.; Chua, K.O.; Lim, P.E. High diversity of bacterial communities in developmental stages of Bactrocera carambolae (Insecta: Tephritidae) revealed by Illumina MiSeq sequencing of 16S rRNA gene. Curr. Microbiol. 2017, 74, 1076–1082. [Google Scholar] [CrossRef]
- Yong, H.S.; Song, S.L.; Chua, K.O.; Lim, P.E. Microbiota associated with Bactrocera carambolae and B. dorsalis (Insecta: Tephritidae) revealed by next-generation sequencing of 16S rRNA gene. Meta Gene 2017, 11, 189–196. [Google Scholar] [CrossRef]
- Mohammed, W.S.; Ziganshina, E.E.; Shagimardanova, E.I.; Gogoleva, N.E.; Ziganshin, A.M. Comparison of intestinal bacterial and fungal communities across various xylophagous beetle larvae (Coleoptera: Cerambycidae). Sci. Rep. 2018, 8, 10073. [Google Scholar] [CrossRef]
- Andongma, A.A.; Wan, L.; Dong, Y.C.; Desneux, N.; White, J.A.; Niu, C.Y. Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis. Sci. Rep. 2015, 5, 9470. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.S.; Song, S.L.; Chua, K.O.; Lim, P.E. Predominance of Wolbachia endosymbiont in the microbiota across life stages of Bactrocera latifrons (Insecta: Tephritidae). Meta Gene 2017, 14, 6–11. [Google Scholar] [CrossRef]
- Malacrinò, A.; Campolo, O.; Medina, R.F.; Palmeri, V. Instar-and host-associated differentiation of bacterial communities in the Mediterranean fruit fly Ceratitis capitata. PLoS ONE 2018, 13, e0194131. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.S.; Shi, G.; Liu, L.J.; Li, Z.H. The diversity of bacteria in different life stages and their impact on the development and reproduction of Zeugodacus tau (Diptera: Tephritidae). Insect Sci. 2020, 00, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Powell, K.S.; Weldon, C.W.; Taylor, P.W. The ecology of Bactrocera tryoni (Diptera: Tephritidae): What do we know to assist pest management? Ann. Appl. Biol. 2011, 158, 26–54. [Google Scholar] [CrossRef] [Green Version]
- Dominiak, B.C.; Mapson, R. Revised distribution of Bactrocera tryoni in eastern Australia and effect on possible incursions of Mediterranean fruit fly: Development of Australia’s eastern trading block. J. Econ. Entomol. 2017, 110, 2459–2465. [Google Scholar] [CrossRef]
- Hancock, D.; Hamacek, E.; Lloyd, A.; Elson-Harris, M. The Distribution and Host Plants of Fruit Flies (Diptera: Tephritidae) in Australia; Queensland Department of Primary Industries: Brisbane, Queensland, Australia, 2000. [Google Scholar]
- Stringer, L.D.; Kean, J.M.; Beggs, J.R.; Suckling, D.M. Management and eradication options for Queensland fruit fly. Popul. Ecol. 2017, 59, 259–273. [Google Scholar] [CrossRef]
- Fletcher, B. The ecology of a natural population of the Queensland fruit fly, Dacus tryoni. V. The dispersal of adults. Aust. J. Zool. 1974, 22, 189–202. [Google Scholar] [CrossRef]
- Gilchrist, A.; Cameron, E.; Sved, J.; Meats, A. Genetic consequences of domestication and mass rearing of pest fruit fly Bactrocera tryoni (Diptera: Tephritidae). J. Econ. Entomol. 2012, 105, 1051–1056. [Google Scholar] [CrossRef]
- Meats, A.; Holmes, H.; Kelly, G. Laboratory adaptation of Bactrocera tryoni (Diptera: Tephritidae) decreases mating age and increases protein consumption and number of eggs produced per milligram of protein. Bull. Entomol. Res. 2004, 94, 517–524. [Google Scholar] [CrossRef]
- Pérez, J.; Park, S.J.; Taylor, P.W. Domestication modifies the volatile emissions produced by male Queensland fruit flies during sexual advertisement. Sci. Rep. 2018, 8, 16503. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.R.; Taylor, P.W. Fecundity, fertility and reproductive recovery of irradiated Queensland fruit fly Bactrocera tryoni. Physiol. Entomol. 2011, 36, 247–252. [Google Scholar] [CrossRef]
- Dominiak, B.; Sundaralingam, S.; Jiang, L.; Jessup, A.; Barchia, I. Quality parameters of mass production adult Queensland fruit fly Bactrocera tryoni (Froggatt) (Diptera: Tephritidae) in 1998/1999. Plant Prot. Q. 2007, 22, 59. [Google Scholar]
- Pérez-Staples, D.; Prabhu, V.; Taylor, P.W. Post-teneral protein feeding enhances sexual performance of Queensland fruit flies. Physiol. Entomol. 2007, 32, 225–232. [Google Scholar] [CrossRef]
- Radhakrishnan, P.; Pérez-Staples, D.; Weldon, C.W.; Taylor, P.W. Multiple mating and sperm depletion in male Queensland fruit flies: Effects on female remating behaviour. Anim. Behav. 2009, 78, 839–846. [Google Scholar] [CrossRef]
- Tychsen, P. Mating behaviour of the Queensland fruit fly, Dacus tryoni (Diptera: Tephritidae) in field cages. J. Aust. Entomol. Soc. 1977, 16, 459–465. [Google Scholar] [CrossRef]
- Vijaysegaran, S.; Walter, G.; Drew, R. Mouthpart structure, feeding mechanisms, and natural food sources of adult Bactrocera (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 1997, 90, 184–201. [Google Scholar] [CrossRef]
- Fanson, B.G.; Taylor, P.W. Protein: Carbohydrate ratios explain life span patterns found in Queensland fruit fly on diets varying in yeast: Sugar ratios. Age 2012, 34, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Fanson, B.G.; Weldon, C.W.; Pérez-Staples, D.; Simpson, S.J.; Taylor, P.W. Nutrients, not caloric restriction, extend lifespan in Queensland fruit flies (Bactrocera tryoni). Aging Cell 2009, 8, 514–523. [Google Scholar] [CrossRef]
- Moadeli, T.; Mainali, B.; Ponton, F.; Taylor, P. Evaluation of yeasts in gel larval diet for Queensland fruit fly, Bactrocera tryoni. J. Appl. Entomol. 2018, 142, 679–688. [Google Scholar] [CrossRef]
- Deutscher, A.T.; Burke, C.M.; Darling, A.E.; Riegler, M.; Reynolds, O.L.; Chapman, T.A. Near full-length 16S rRNA gene next-generation sequencing revealed Asaia as a common midgut bacterium of wild and domesticated Queensland fruit fly larvae. Microbiome 2018, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, R.; Sutcliffe, B.; Taylor, P.W.; Chapman, T.A. Next-Generation Sequencing reveals relationship between the larval microbiome and food substrate in the polyphagous Queensland fruit fly. Sci. Rep. 2019, 9, 14292. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.L.; Frommer, M.; Shearman, D.C.; Riegler, M. The microbiome of field-caught and laboratory-adapted Australian tephritid fruit fly species with different host plant use and specialisation. Microb. Ecol. 2015, 70, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Thaochan, N.; Drew, R.; Hughes, J.; Vijaysegaran, S.; Chinajariyawong, A. Alimentary tract bacteria isolated and identified with API-20E and molecular cloning techniques from Australian tropical fruit flies, Bactrocera cacuminata and B. tryoni. J. Insect Sci. 2010, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Woruba, D.N.; Morrow, J.L.; Reynolds, O.L.; Chapman, T.A.; Collins, D.P.; Riegler, M. Diet and irradiation effects on the bacterial community composition and structure in the gut of domesticated teneral and mature Queensland fruit fly, Bactrocera tryoni (Diptera: Tephritidae). BMC Microbiol. 2019, 19, 281. [Google Scholar] [CrossRef] [Green Version]
- Fitt, G.P.; O’Brien, R. Bacteria associated with four species of Dacus (Diptera: Tephritidae) and their role in the nutrition of the larvae. Oecologia 1985, 67, 447–454. [Google Scholar] [CrossRef]
- Deutscher, A.T.; Reynolds, O.L.; Chapman, T.A. Yeast: An overlooked component of Bactrocera tryoni (Diptera: Tephritidae) larval gut microbiota. J. Econ. Entomol. 2016, 110, 298–300. [Google Scholar] [CrossRef]
- Piper, A.M.; Farnier, K.; Linder, T.; Speight, R.; Cunningham, J.P. Two gut-associated yeasts in a Tephritid fruit fly have contrasting effects on adult attraction and larval survival. J. Chem. Ecol. 2017, 43, 891–901. [Google Scholar] [CrossRef]
- Andongma, A.A.; Wan, L.; Dong, Y.C.; Wang, Y.L.; He, J.; Niu, C.Y. Assessment of the Bacteria community structure across life stages of the Chinese Citrus Fly, Bactrocera minax (Diptera: Tephritidae). BMC Microbiol. 2019, 19, 285. [Google Scholar] [CrossRef] [Green Version]
- Morales-Jiménez, J.; Zúñiga, G.; Ramírez-Saad, H.C.; Hernández-Rodríguez, C. Gut-associated bacteria throughout the life cycle of the bark beetle Dendroctonus rhizophagus Thomas and Bright (Curculionidae: Scolytinae) and their cellulolytic activities. Microb. Ecol. 2012, 64, 268–278. [Google Scholar] [CrossRef]
- Hammer, T.J.; McMillan, W.O.; Fierer, N. Metamorphosis of a butterfly-associated bacterial community. PLoS ONE 2014, 9, e86995. [Google Scholar] [CrossRef]
- Moll, R.M.; Romoser, W.S.; Modrakowski, M.C.; Moncayo, A.C.; Lerdthusnee, K. Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J. Med. Entomol. 2001, 38, 29–32. [Google Scholar] [CrossRef]
- Martinson, V.G.; Douglas, A.E.; Jaenike, J. Community structure of the gut microbiota in sympatric species of wild Drosophila. Ecol. Lett. 2017, 20, 629–639. [Google Scholar] [CrossRef]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.D.; Kim, Y.J.; Choi, J.H. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulou, P.; Asimakis, E.D.; Khan, M.; Caceres, C.; Bourtzis, K.; Tsiamis, G. Irradiation effect on the structure of bacterial communities associated with the oriental fruit fly, Bactrocera dorsalis. Entomol. Exp. Appl. 2019, 167, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, X.; Chen, Z.; Wang, Z.; Lu, Y.; Cheng, D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Yao, Z.; Zheng, W.; Zhang, H. Bacterial communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 2014, 9, e106988. [Google Scholar] [CrossRef]
- Behar, A.; Jurkevitch, E.; Yuval, B. Bringing back the fruit into fruit fly–bacteria interactions. Mol. Ecol. 2008, 17, 1375–1386. [Google Scholar] [CrossRef]
- Ben-Yosef, M.; Pasternak, Z.; Jurkevitch, E.; Yuval, B. Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. J. Evol. Biol. 2014, 27, 2695–2705. [Google Scholar] [CrossRef]
- Augustinos, A.A.; Kyritsis, G.A.; Papadopoulos, N.T.; Abd-Alla, A.M.; Cáceres, C.; Bourtzis, K. Exploitation of the medfly gut microbiota for the enhancement of sterile insect technique: Use of Enterobacter sp. in larval diet-based probiotic applications. PLoS ONE 2015, 10, e0136459. [Google Scholar] [CrossRef] [Green Version]
- Ben Ami, E.; Yuval, B.; Jurkevitch, E. Manipulation of the microbiota of mass-reared Mediterranean fruit flies Ceratitis capitata (Diptera: Tephritidae) improves sterile male sexual performance. ISME J. 2010, 4, 28–37. [Google Scholar]
- Gavriel, S.; Jurkevitch, E.; Gazit, Y.; Yuval, B. Bacterially enriched diet improves sexual performance of sterile male Mediterranean fruit flies. J. Appl. Entomol. 2011, 135, 564–573. [Google Scholar] [CrossRef]
- Hamden, H.; Guerfali, M.M.S.; Fadhl, S.; Saidi, M.; Chevrier, C. Fitness improvement of mass-reared sterile males of Ceratitis capitata (Vienna 8 strain) (Diptera: Tephritidae) after gut enrichment with probiotics. J. Econ. Entomol. 2013, 106, 641–647. [Google Scholar] [CrossRef]
- Martínez-Falcón, A.P.; Durbán, A.; Latorre, A.; Antón, J.; de los Ángeles Marcos-García, M. Bacteria associated with Copestylum (Diptera, Syrphidae) larvae and their cactus host Isolatocereus dumortieri. PLoS ONE 2011, 6, e27443. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, P.; Granchietti, A.; Landini, S.; Viti, C.; Giovannetti, L.; Belcari, A. Relationships between the olive fly and bacteria. J. Appl. Entomol. 2008, 132, 682–689. [Google Scholar] [CrossRef]
- Crotti, E.; Rizzi, A.; Chouaia, B.; Ricci, I.; Favia, G.; Alma, A.; Sacchi, L.; Bourtzis, K.; Mandrioli, M.; Cherif, A. Acetic acid bacteria, newly emerging symbionts of insects. Appl. Environ. Microbiol. 2010, 76, 6963–6970. [Google Scholar] [CrossRef] [Green Version]
- Chouaia, B.; Rossi, P.; Montagna, M.; Ricci, I.; Crotti, E.; Damiani, C.; Epis, S.; Faye, I.; Sagnon, N.F.; Alma, A. Molecular evidence for multiple infections as revealed by typing of Asaia bacterial symbionts of four mosquito species. Appl. Environ. Microbiol. 2010, 76, 7444–7450. [Google Scholar] [CrossRef] [Green Version]
- Mitraka, E.; Stathopoulos, S.; Siden-Kiamos, I.; Christophides, G.K.; Louis, C. Asaia accelerates larval development of Anopheles gambiae. Pathog. Glob. Health 2013, 107, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Kiss, L. Limits of nuclear ribosomal DNA internal transcribed spacer (ITS) sequences as species barcodes for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, E1811. [Google Scholar] [CrossRef] [Green Version]
- Malacrinò, A.; Schena, L.; Campolo, O.; Laudani, F.; Palmeri, V. Molecular analysis of the fungal microbiome associated with the olive fruit fly Bactrocera oleae. Fungal Ecol. 2015, 18, 67–74. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.M. Invertebrate-Microbial Interactions. Ingested Fungal Enzymes in Arthropod Biology; Cornell University Press: Ithaca, NY, USA, 1987. [Google Scholar]
- Nardon, P.; Grenier, A. Endocytobiosis in Coleoptera: Biological, biochemical, and genetic aspects. Insect Endocytobiosis 1989, 11, 175–215. [Google Scholar]
- Carvalho, M.; Schwudke, D.; Sampaio, J.L.; Palm, W.; Riezman, I.; Dey, G.; Gupta, G.D.; Mayor, S.; Riezman, H.; Shevchenko, A. Survival strategies of a sterol auxotroph. Development 2010, 137, 3675–3685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jiao, S.; Li, X.; Li, M. Bacterial and fungal gut communities of Agrilus mali at different developmental stages and fed different diets. Sci. Rep. 2018, 8, 15634. [Google Scholar] [CrossRef] [Green Version]
- Franzini, P.Z.; Ramond, J.-B.; Scholtz, C.H.; Sole, C.L.; Ronca, S.; Cowan, D.A. The gut microbiomes of two Pachysoma MacLeay desert dung beetle species (Coleoptera: Scarabaeidae: Scarabaeinae) feeding on different diets. PLoS ONE 2016, 11, e0161118. [Google Scholar]
- Hu, X.; Li, M.; Chen, H. Community structure of gut fungi during different developmental stages of the Chinese white pine beetle (Dendroctonus armandi). Sci. Rep. 2015, 5, 8411. [Google Scholar] [CrossRef]
- Hamby, K.A.; Hernández, A.; Boundy-Mills, K.; Zalom, F.G. Associations of yeasts with spotted-wing Drosophila (Drosophila suzukii; Diptera: Drosophilidae) in cherries and raspberries. Appl. Environ. Microbiol. 2012, 78, 4869–4873. [Google Scholar] [CrossRef] [Green Version]
- El Haidani, A.; Chakri, M.; Mostakim, M.; El Mzibri, M.; Boudouma, J.; El Hassouni, M.; Haggoud, A.; Iraqui, M.; Houari, A.; Ibnsouda, S.K. Isolation and characterisation of yeast strains for the olive fly Bactrocera oleae biological control. Moroc. J. Biol. 2008, 2, 19–29. [Google Scholar]
- Konstantopoulou, M.; Mazomenos, B. Evaluation of Beauveria bassiana and B. brongniartii strains and four wild-type fungal species against adults of Bactrocera oleae and Ceratitis capitata. BioControl 2005, 50, 293–305. [Google Scholar] [CrossRef]
- Demain, A.L.; Fang, A. The natural functions of secondary metabolites. In History of Modern Biotechnology I; Springer: Berlin/Heidelberg, Germany, 2000; pp. 1–39. [Google Scholar]
- Sweeney, M.J.; Dobson, A.D. Mycotoxin production by Aspergillus, Fusarium and Penicillium species. Int. J. Food Microbiol. 1998, 43, 141–158. [Google Scholar] [CrossRef]
- Flessa, F.; Peršoh, D.; Rambold, G. Annuality of Central European deciduous tree leaves delimits community development of epifoliar pigmented fungi. Fungal Ecol. 2012, 5, 554–561. [Google Scholar] [CrossRef]
- Frisullo, S.; Carlucci, A. Minor fungal diseases of olive. In Olive Diseases and Disorders; Schena, L., Agosteo, G., Cacciola, S.O., Eds.; Transworld Research Network: Kerala, India, 2011; pp. 209–304. [Google Scholar]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus cladosporium. Stud. Mycol. 2012, 72, 1–401. [Google Scholar] [CrossRef] [Green Version]
- Swett, C.L.; Hamby, K.A.; Hellman, E.M.; Carignan, C.; Bourret, T.B.; Koivunen, E.E. Characterizing members of the Cladosporium cladosporioides species complex as fruit rot pathogens of red raspberries in the mid-Atlantic and co-occurrence with Drosophila suzukii (spotted wing drosophila). Phytoparasitica 2019, 47, 415–428. [Google Scholar] [CrossRef]
- Sung-Oui, S.; Mchugh, J.V.; Pollock, D.D.; Blackwell, M. The beetle gut: A hyperdiverse source of novel yeasts. Mycol. Res. 2005, 109, 261–265. [Google Scholar]
- Vogel, H.; Shukla, S.P.; Engl, T.; Weiss, B.; Fischer, R.; Steiger, S.; Heckel, D.G.; Kaltenpoth, M.; Vilcinskas, A. The digestive and defensive basis of carcass utilization by the burying beetle and its microbiota. Nat. Commun. 2017, 8, 15186. [Google Scholar] [CrossRef] [Green Version]
- Kaltenpoth, M.; Steiger, S. Unearthing carrion beetles’ microbiome: Characterization of bacterial and fungal hindgut communities across the S ilphidae. Mol. Ecol. 2014, 23, 1251–1267. [Google Scholar] [CrossRef]
- Cacciola, S.; Faedda, R.; Sinatra, F.; Agosteo, G.; Schena, L.; Frisullo, S.; di San Lio, G.M. Olive anthracnose. J. Plant Pathol. 2012, 94, 29–44. [Google Scholar]
- Schena, L.; Mosca, S.; Cacciola, S.O.; Faedda, R.; Sanzani, S.M.; Agosteo, G.E.; Sergeeva, V.; Magnano di San Lio, G. Species of the Colletotrichum gloeosporioides and C. boninense complexes associated with olive anthracnose. Plant Pathol. 2014, 63, 437–446. [Google Scholar] [CrossRef]
- Moral, J.; de Oliveira, R.; Trapero, A. Elucidation of the disease cycle of olive anthracnose caused by Colletotrichum acutatum. Phytopathology 2009, 99, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Folmer, o.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- White, I.M.; Elson-Harris, M.M. Fruit Flies of Economic Significance: Their Identification and Bionomics; CAB International: Wallingford, UK, 1992. [Google Scholar]
- Hoggard, M.; Vesty, A.; Wong, G.; Montgomery, J.M.; Fourie, C.; Douglas, R.G.; Biswas, K.; Taylor, M.W. Characterizing the human mycobiota: A comparison of small subunit rRNA, ITS1, ITS2, and large subunit rRNA genomic targets. Front. Microbiol. 2018, 9, 2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.A.; Bálint, M.; Greshake, B.; Bandow, C.; Römbke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 2013, 65, 128–132. [Google Scholar] [CrossRef]
- Sutcliffe, B.; Chariton, A.A.; Harford, A.J.; Hose, G.C.; Greenfield, P.; Midgley, D.J.; Paulsen, I.T. Diverse fungal lineages in subtropical ponds are altered by sediment-bound copper. Fungal Ecol. 2018, 34, 28–42. [Google Scholar] [CrossRef]
- Fouts, D.E.; Szpakowski, S.; Purushe, J.; Torralba, M.; Waterman, R.C.; MacNeil, M.D.; Alexander, L.J.; Nelson, K.E. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS ONE 2012, 7, e48289. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Ainsworth, M. A method of linking multivariate community structure to environmental variables. Mar. Ecol. Prog. Ser. 1993, 92, 205. [Google Scholar] [CrossRef]
- Sutcliffe, B.; Chariton, A.A.; Harford, A.J.; Hose, G.C.; Greenfield, P.; Elbourne, L.D.; Oytam, Y.; Stephenson, S.; Midgley, D.J.; Paulsen, I.T. Effects of uranium concentration on microbial community structure and functional potential. Environ. Microbiol. 2017, 19, 3323–3341. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Phylum | Class | Order | Family | Genus | Larvae | Pupae | Adult Male | Adult Female |
|---|---|---|---|---|---|---|---|---|---|
| Bacteria | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | 27.0% | 38.1% | 80.4% | 94.8% | |
| Bacteria | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Acetobacteraceae | Swaminathania/Asaia | 29.2% | 37.6% | 0.2% | 1.1% |
| Bacteria | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Erwinia | 22.6% | 18.1% | 0.0% | 1.1% |
| Bacteria | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Providencia | 0.00% | 0.00% | 19.4% | 2.8% |
| Bacteria | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Acetobacteraceae | Acetobacter | 10.2% | 1.4% | 0.0% | 0.0% |
| Bacteria | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Acetobacteraceae | Gluconobacter | 5.3% | 3.4% | 0.0% | 0.0% |
| Bacteria | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Acetobacteraceae | 3.8% | 0.6% | 0.0% | 0.0% | |
| Bacteria | Bacteroidetes | Flavobacteriia | Flavobacteriales | Weeksellaceae | Wautersiella | 1.7% | 0.0% | 0.0% | 0.0% |
| Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | 0.0% | 0.4% | 0.0% | 0.0% | |
| Bacteria | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | 0.0% | 0.3% | 0.0% | 0.0% | |
| Bacteria | 0.1% | 0.1% | 0.0% | 0.0% |
| Domain | Phylum | Class | Order | Family | Genus | Larvae | Pupae | Adult Male | Adult Female |
|---|---|---|---|---|---|---|---|---|---|
| Fungi | Ascomycota | Eurotiomycetes | Eurotiales | Trichocomaceae | Penicillium | 0.4% | 23.2% | 47.8% | 10.8% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Incertae-sedis | Candida | 38.3% | 21.5% | 0.0% | 0.4% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Pichiaceae | Pichia | 26.5% | 12.1% | 0.1% | 0.1% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Incertae-sedis | Cyberlindnera | 0.0% | 0.0% | 0.0% | 32.3% |
| Fungi | Ascomycota | Sordariomycetes | Hypocreales | Nectriaceae | Gibberella | 0.0% | 0.0% | 19.6% | 0.1% |
| Fungi | Basidiomycota | Tremellomycetes | 0.0% | 0.0% | 19.4% | 0.0% | |||
| Fungi | Ascomycota | Dothideomycetes | Capnodiales | Cladosporiaceae | Cladosporium | 0.0% | 2.9% | 0.0% | 16.3% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetaceae | Zygosaccharomyces | 16.1% | 0.0% | 0.0% | 0.0% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Trichomonascaceae | Zygoascus | 0.0% | 9.2% | 0.3% | 1.8% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Debaryomycetaceae | Meyerozyma | 0.0% | 6.2% | 0.0% | 0.1% |
| Fungi | Ascomycota | Dothideomycetes | Pleosporales | Didymosphaeriaceae | Pseudopithomyces | 0.0% | 0.0% | 4.1% | 0.0% |
| Fungi | Ascomycota | Eurotiomycetes | Eurotiales | Trichocomaceae | Aspergillus | 0.0% | 3.3% | 0.0% | 0.0% |
| Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetaceae | Saccharomyces | 0.0% | 0.0% | 0.0% | 3.3% |
| Fungi | 18.2% | 20.1% | 8.6% | 33.3% |
| Geographic Location of Collection | Fruit Source and Number of Fruits Collected | Collection Date |
|---|---|---|
| Coomealla, NSW GPS: Lat 34° 5¡ä50.97", Long 142° 3¡ä7.21" | Pomegranate 37 pieces | 5/05/17 |
| St. Germains, Between Tatura and Echuca in Victoria GPS: Lat 36°10’48.86", Long 145° 8’50.74" | Green Apple 41 pieces | 05/05/17 |
| Downer road between Tatura and Toolamba in Victoria GPS: Lat 26°38’34.92", Long 152°56’22.99" | Quince 52 pieces | 05/05/17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majumder, R.; Sutcliffe, B.; Taylor, P.W.; Chapman, T.A. Microbiome of the Queensland Fruit Fly through Metamorphosis. Microorganisms 2020, 8, 795. https://doi.org/10.3390/microorganisms8060795
Majumder R, Sutcliffe B, Taylor PW, Chapman TA. Microbiome of the Queensland Fruit Fly through Metamorphosis. Microorganisms. 2020; 8(6):795. https://doi.org/10.3390/microorganisms8060795
Chicago/Turabian StyleMajumder, Rajib, Brodie Sutcliffe, Phillip W. Taylor, and Toni A. Chapman. 2020. "Microbiome of the Queensland Fruit Fly through Metamorphosis" Microorganisms 8, no. 6: 795. https://doi.org/10.3390/microorganisms8060795
APA StyleMajumder, R., Sutcliffe, B., Taylor, P. W., & Chapman, T. A. (2020). Microbiome of the Queensland Fruit Fly through Metamorphosis. Microorganisms, 8(6), 795. https://doi.org/10.3390/microorganisms8060795