Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing, Read Alignment and Genotype Calling
2.2. Phylogenetic Analyses and Survey of Specific Genes
2.3. Phenotypic Analyses
3. Results
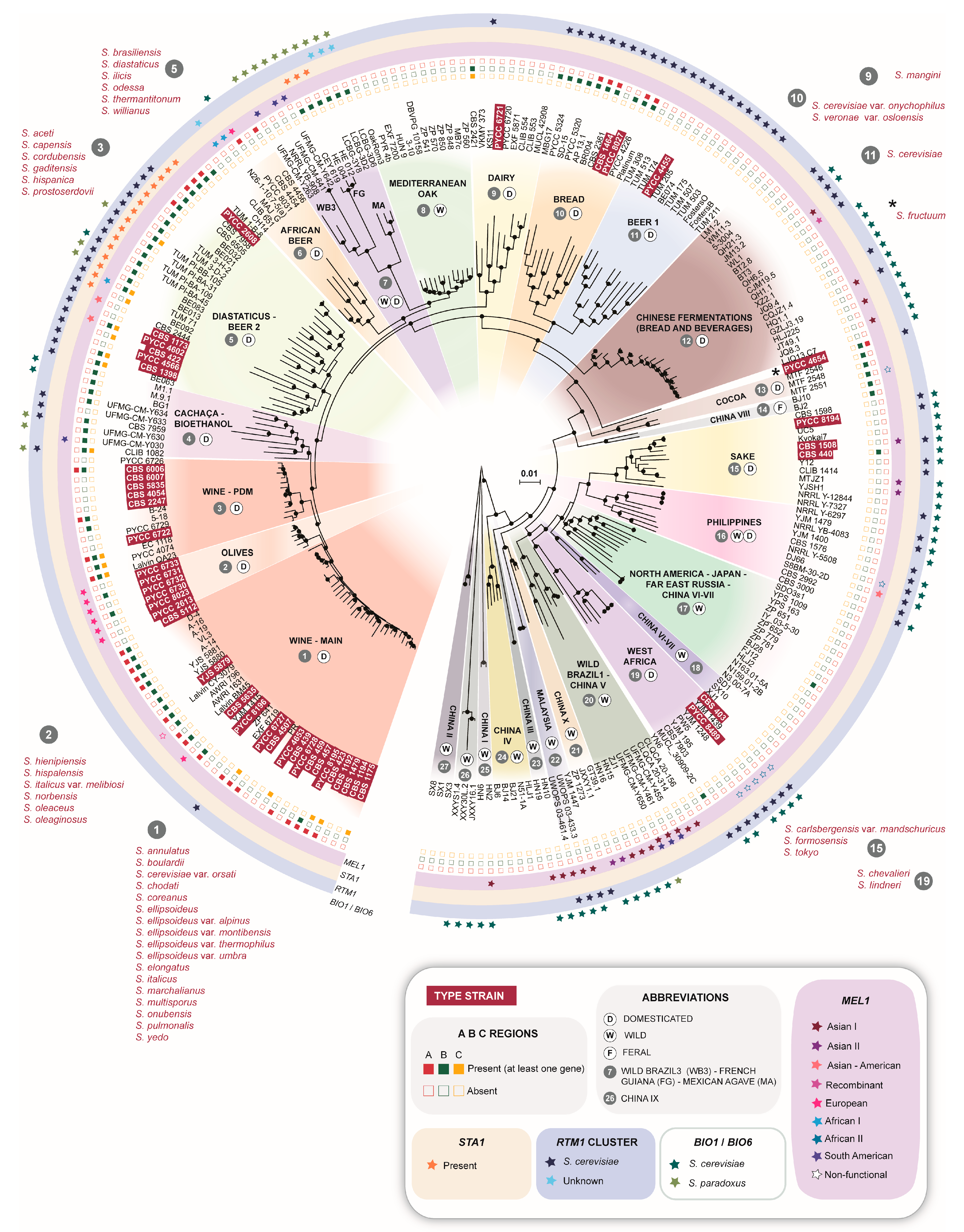
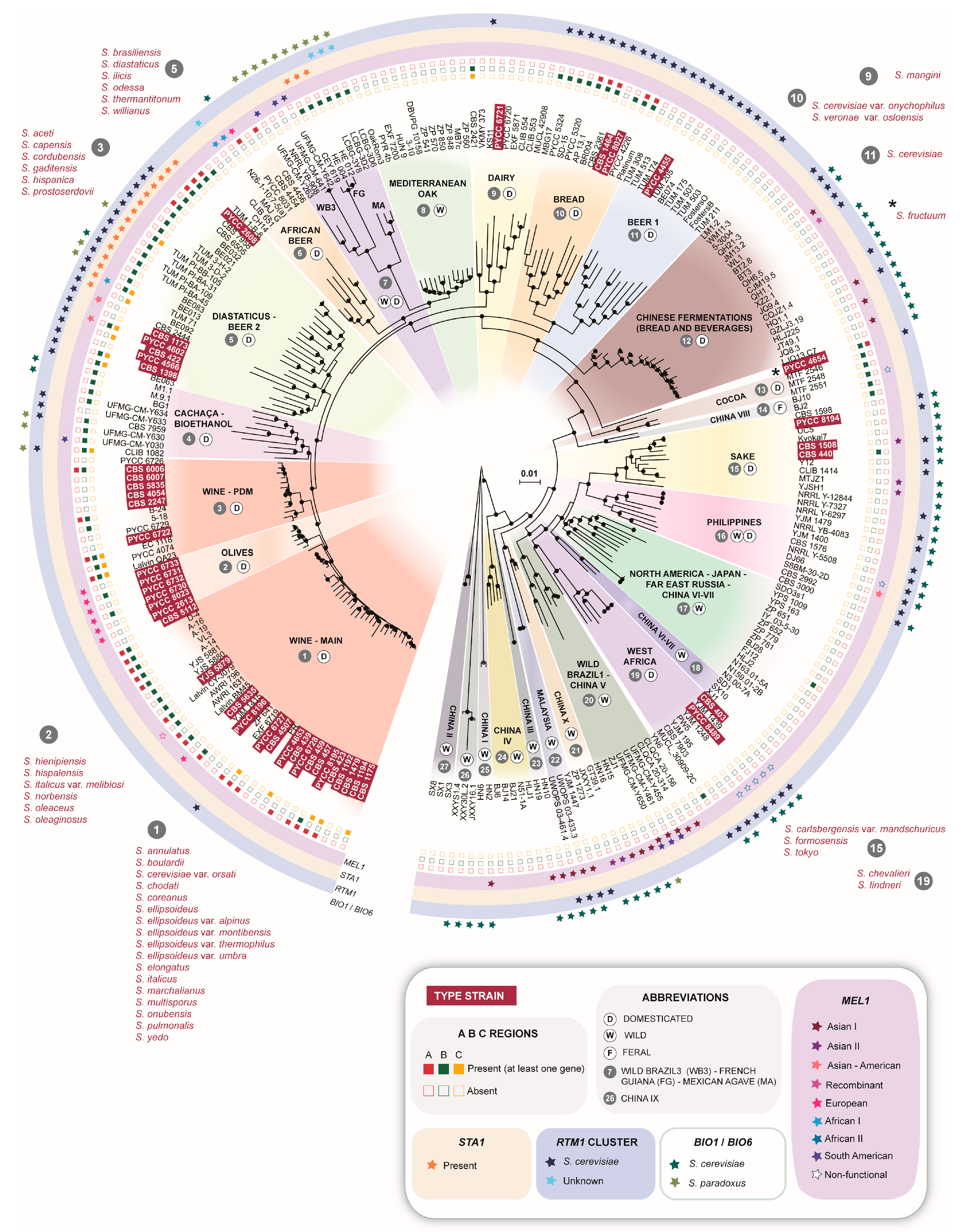
3.1. Global Phylogenetic Analysis Reveals an Unequal Distribution of Type Strains of Former Species
3.2. The Wine and Olives Clades Harbor Most Type Strains of Former Species
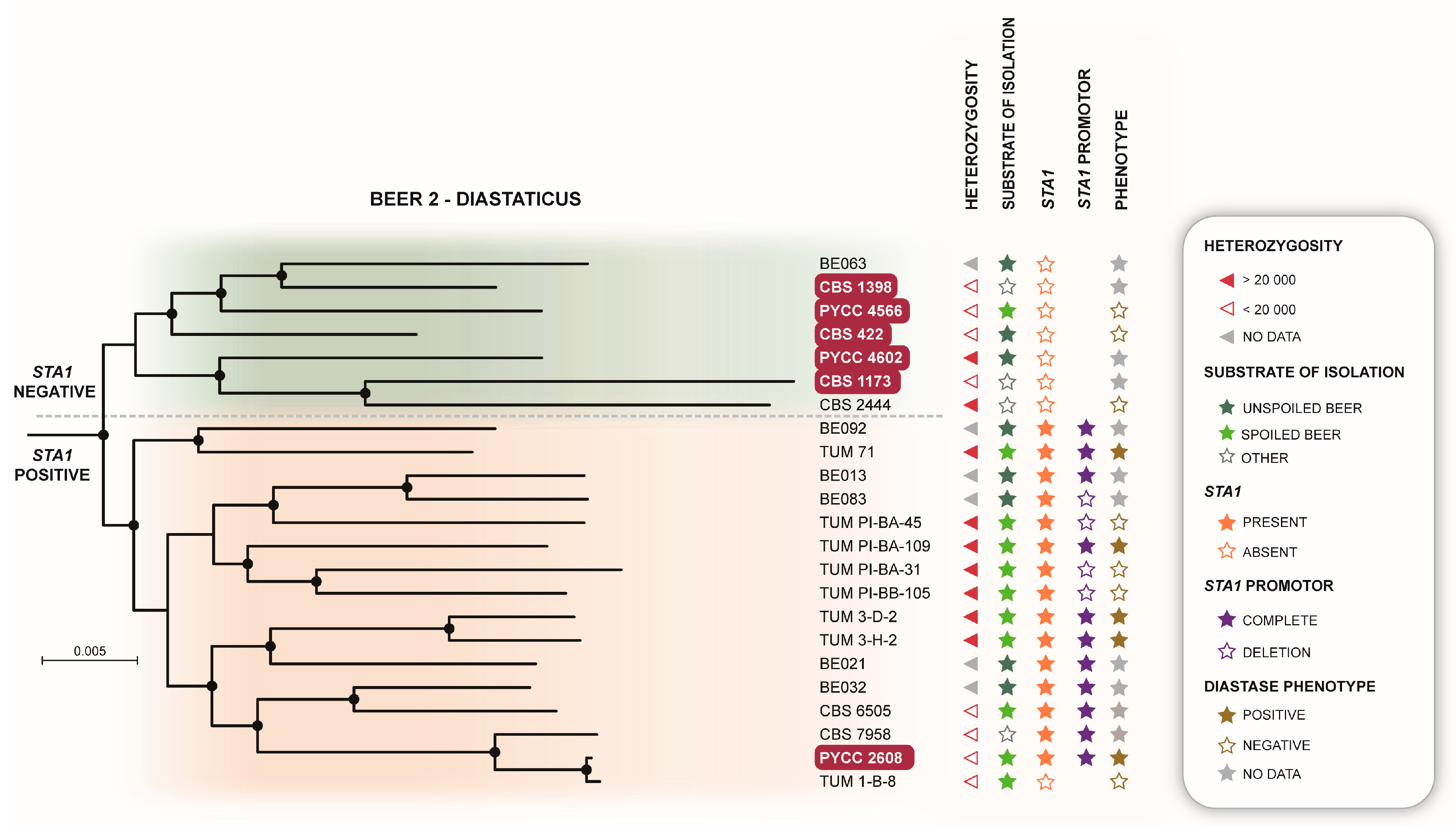
3.3. The Beer and Bread Clades Contain Multiple Type Strains of Former Species
3.4. Additional Clades Harboring Type Strains of Former Species
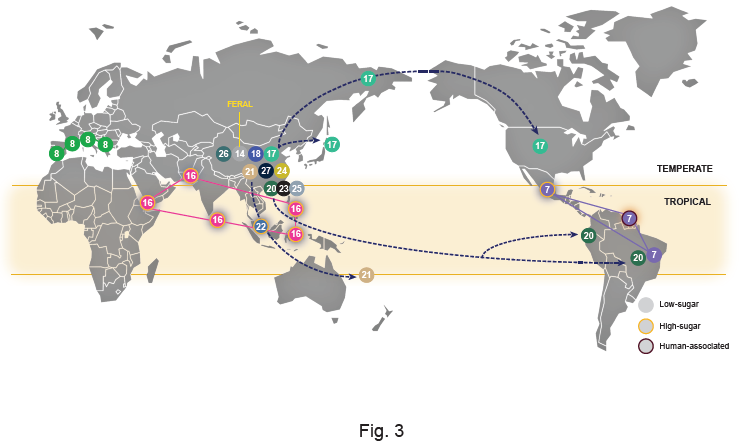
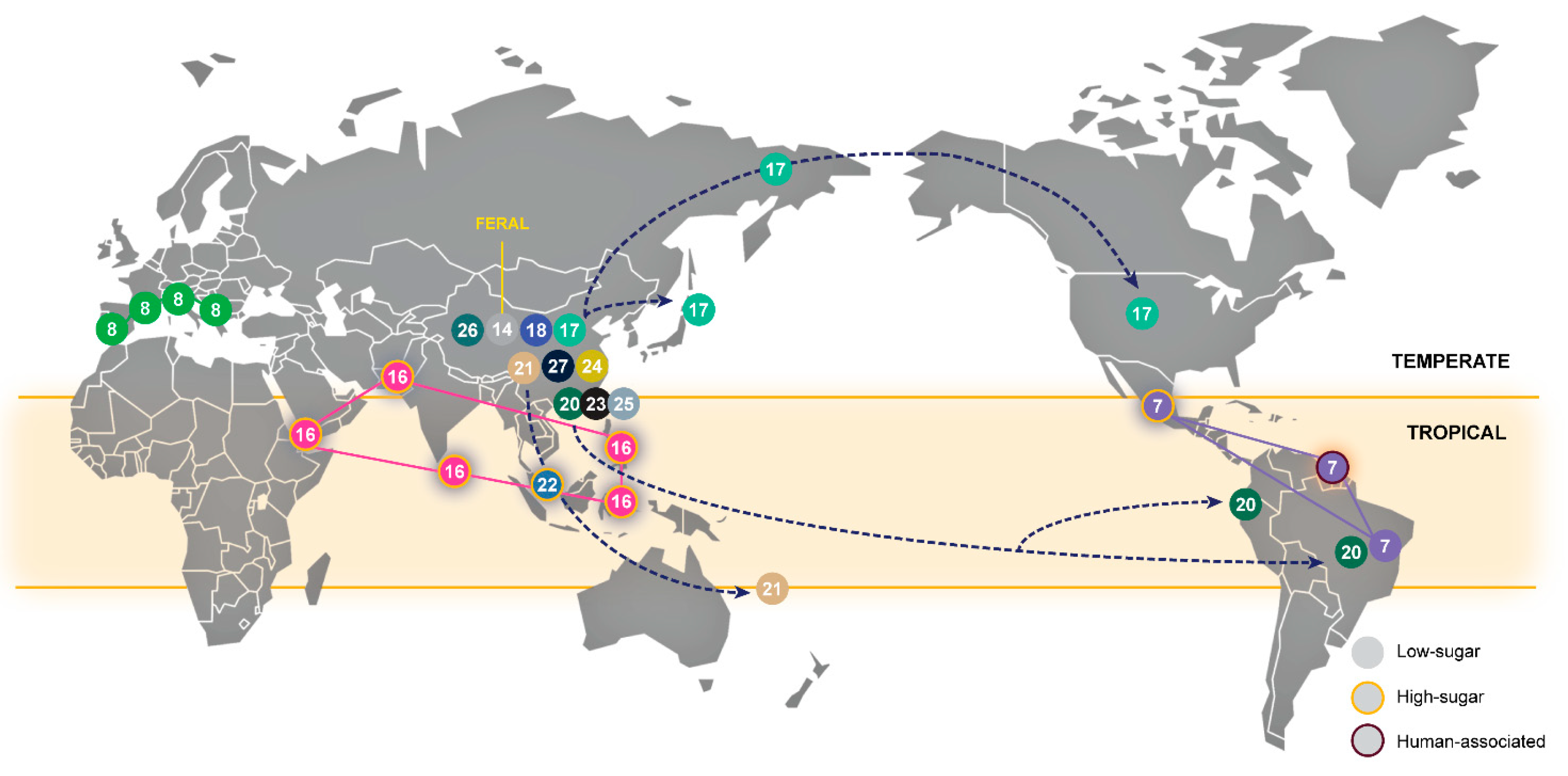
3.5. The Biogeography and Ecology of Wild Populations
3.6. Lifestyle Drives the Fate of Aquaporins
3.7. The Natural Reservoirs of RTM1 and BIO1/BIO6
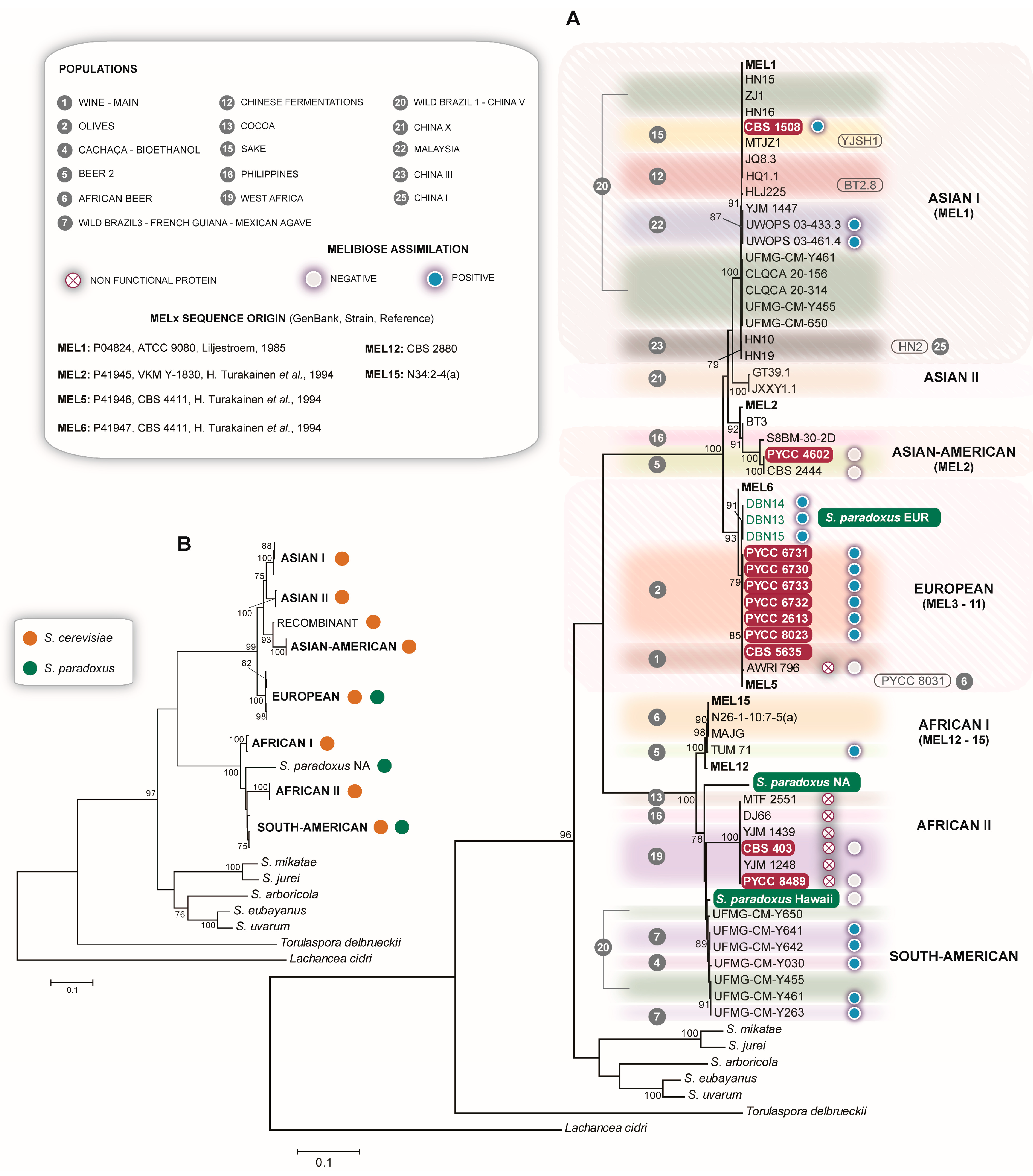
3.8. The Complex Distribution of MEL Alleles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meyen, J. Jahresbericht uber die Resultate der Arbeiten im Felde der physiologischen Botanik von dem Jahre 1837. Arch. Naturgesch. 1838, 4, 1–186. [Google Scholar]
- Stelling-Dekker, N.M. Die sporogenen Hefen. Verh. K. Ned. Akad. Wetensch. Afd. Natuurk. Sect. II 1931, 28, 1–547. [Google Scholar]
- Lodder, J.; Kreger van Rij, N. The Yeasts, A Taxonomic Study; North-Holland: Amsterdam, The Netherlands, 1952. [Google Scholar]
- Van der Walt, J.P. Saccharomyces (Meyen) emend. Reess. In The Yeasts, A Taxonomic Study; Lodder, J., Ed.; North-Holland: Amsterdam, The Netherlands, 1970; pp. 555–718. [Google Scholar]
- Yarrow, D. Saccharomyces Meyen ex Reess. In The Yeasts, A Taxonomic Study; Kreger van Rig, N., Ed.; Elsevier: Amsterdam, The Netherlands, 1984; pp. 379–395. [Google Scholar]
- Liti, G.; Carter, D.M.; Moses, A.M.; Warringer, J.; Parts, L.; James, S.A.; Davey, R.P.; Roberts, I.N.; Burt, A.; Koufopanou, V.; et al. Population genomics of domestic and wild yeasts. Nature 2009, 458, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strope, P.K.; Skelly, D.A.; Kozmin, S.G.; Mahadevan, G.; Stone, E.A.; Magwene, P.M.; Dietrich, F.S.; McCusker, J.H. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 2015, 25, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.; Barbosa, R.; Zalar, P.; Imanishi, Y.; Shimizu, K.; Turchetti, B.; Legras, J.-L.; Serra, M.; Dequin, S.; Couloux, A.; et al. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol. Ecol. 2015, 24, 5412–5427. [Google Scholar] [CrossRef]
- Peter, J.; de Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barré, B.P.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Scannell, D.R.; Zill, O.; Rokas, A.; Payen, C.; Dunham, M.J.; Eisen, M.B.; Rine, J.; Johnston, M.; Hittinger, C.T. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 Genes/Genomes/Genetics 2011, 1, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Liti, G.; Ba, A.N.N.; Blythe, M.J.; Müller, C.A.; Bergström, A.; Cubillos, F.A.; Dafhnis-Calas, F.; Khoshraftar, S.; Malla, S.; Mehta, N.; et al. High quality de novo sequencing and assembly of the Saccharomyces arboricolus genome. BMC Genom. 2013, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- Borneman, A.R.; Forgan, A.H.; Kolouchova, R.; Fraser, J.A.; Schmidt, S.A. Whole genome comparison reveals high levels of inbreeding and strain redundancy across the spectrum of commercial wine strains of Saccharomyces cerevisiae. G3 Genes/Genomes/Genetics 2016, 6, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.R.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Almeida, P.; Gonçalves, C.; Teixeira, S.; Libkind, D.; Bontrager, M.; Masneuf-Pomarede, I.; Albertin, W.; Durrens, P.; Sherman, D.J.; Marullo, P.; et al. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat. Commun. 2014, 5, 4044. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Boil. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, J.L.; Kim, H.S.; Clarke, J.; Painter, J.C.; Fay, J.C.; Gasch, A.P. Incipient balancing selection through adaptive loss of aquaporins in natural Saccharomyces cerevisiae populations. PLoS Genet. 2010, 6, e1000893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, M.; Bigey, F.; Beyne, E.; Galeote, V.; Gavory, F.; Mallet, S.; Cambon, B.; Legras, J.-L.; Wincker, P.; Casaregola, S.; et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc. Natl. Acad. Sci. USA 2009, 106, 16333–16338. [Google Scholar] [CrossRef] [Green Version]
- Nijkamp, J.; Broek, M.V.D.; Datema, E.; de Kok, S.; Bosman, L.; Luttik, M.A.; Daran-Lapujade, P.; Vongsangnak, W.; Nielsen, J.; Heijne, W.H.; et al. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb. Cell Factories 2012, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Warringer, J.; Zörgö, E.; Cubillos, F.A.; Zia, A.; Gjuvsland, A.B.; Simpson, J.T.; Forsmark, A.; Durbin, R.; Omholt, S.W.; Louis, E.; et al. Trait variation in yeast is defined by population history. PLoS Genet. 2011, 7, e1002111. [Google Scholar] [CrossRef] [Green Version]
- Krogerus, K.; Magalhães, F.; Kuivanen, J.; Gibson, B. A deletion in the STA1 promoter determines maltotriose and starch utilization in STA1+ Saccharomyces cerevisiae strains. Appl. Microbiol. Biotechnol. 2019, 103, 7597–7615. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.-X.; Li, J.; Aigrain, L.; Hallin, J.; Persson, K.; Oliver, K.; Bergström, A.; Coupland, P.; Warringer, J.; Lagomarsino, M.C.; et al. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat. Genet. 2017, 49, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legras, J.-L.; Galeote, V.; Bigey, F.; Camarasa, C.; Marsit, S.; Nidelet, T.; Sanchez, I.; Couloux, A.; Guy, J.; Franco-Duarte, R.; et al. Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Mol. Biol. Evol. 2018, 35, 1712–1727. [Google Scholar] [CrossRef] [PubMed]
- Pontes, A.; Čadež, N.; Gonçalves, P.; Sampaio, J.P. A quasi-domesticate relic hybrid population of Saccharomyces cerevisiae × S. paradoxus adapted to olive brine. Front. Genet. 2019, 10, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromie, G.A.; Hyma, K.E.; Ludlow, C.L.; Garmendia-Torres, C.; Gilbert, T.L.; May, P.; Huang, A.A.; Dudley, A.M.; Fay, J.C. Genomic sequence diversity and population structure of Saccharomyces cerevisiae assessed by RAD-seq. G3 (Bethesda) 2013, 3, 2163–2171. [Google Scholar] [CrossRef] [Green Version]
- Legras, J.L.; Merdinoglu, D.; Cornuet, J.M.; Karst, F. Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Mol. Ecol. 2007, 16, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, R.; Pontes, A.; Santos, R.O.; Montandon, G.G.; de Ponzzes-Gomes, C.M.; Morais, P.B.; Gonçalves, P.; Rosa, C.A.; Sampaio, J.P. Multiple rounds of artificial selection promote microbe secondary domestication—the case of cachaça yeasts. Genome Biol. Evol. 2018, 10, 1939–1955. [Google Scholar] [CrossRef] [PubMed]
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L.; et al. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 2016, 166, 1397–1410. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, R.; Almeida, P.; Safar, S.V.B.; Santos, R.O.; Morais, P.B.; Nielly-Thibault, L.; Leducq, J.-B.; Landry, C.R.; Gonçalves, P.; Rosa, C.A.; et al. Evidence of natural hybridization in Brazilian wild lineages of Saccharomyces cerevisiae. Genome Biol. Evol. 2016, 8, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Angebault, C.; Djossou, F.; Abélanet, S.; Permal, E.; Soltana, M.B.; Diancourt, L.; Bouchier, C.; Woerther, P.L.; Catzeflis, F.; Andremont, A.; et al. Candida albicans is not always the preferential yeast colonizing humans: a study inwayampi amerindians. J. Infect. Dis. 2013, 208, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Han, P.-J.; Wang, Q.-M.; Liu, W.-Q.; Shi, J.-Y.; Li, K.; Zhang, X.-L.; Bai, F.-Y. The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat. Commun. 2018, 9, 2690. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-M.; Liu, W.-Q.; Liti, G.; Wang, S.-A.; Bai, F.-Y. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol. Ecol. 2012, 21, 5404–5417. [Google Scholar] [CrossRef] [PubMed]
- Zanello, G.; Meurens, F.; Berri, M.; Salmon, H. Saccharomyces boulardii effects on gastrointestinal diseases. Curr. Issues Mol. Biol. 2008, 11, 47–58. [Google Scholar] [PubMed]
- Legras, J.-L.; Erny, C.; Charpentier, C. Population structure and comparative genome hybridization of European flor yeast reveal a unique group of Saccharomyces cerevisiae strains with few gene duplications in their genome. PLoS ONE 2014, 9, e108089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torresi, S.; Frangipane, M.T.; Anelli, G. Biotechnologies in sparkling wine production. Interesting approaches for quality improvement: A review. Food Chem. 2011, 129, 1232–1241. [Google Scholar] [CrossRef]
- Marsit, S.; Leducq, J.-B.; Durand, E.; Marchant, A.; Filteau, M.; Landry, C.R. Evolutionary biology through the lens of budding yeast comparative genomics. Nat. Rev. Genet. 2017, 18, 581–598. [Google Scholar] [CrossRef]
- Andrews, B.J.; Gilliland, R.B. Super-attenuation of beer: A study of three organisms capable of causing abnormal attenuations. J. Inst. Brew. 1952, 58, 189–196. [Google Scholar] [CrossRef]
- Van der Walt, J.P. Saccharomyces vafer and S. inconspicuus spp.n. Antonie Leeuwenhoek 1965, 31, 187–192. [Google Scholar] [CrossRef]
- Gilliland, R.B. Sacchatomyces diastaticus—A starch-fermenting yeast. J. Inst. Brew. 1966, 72, 271–275. [Google Scholar] [CrossRef]
- Meier-Dörnberg, T.; Kory, O.I.; Jacob, F.; Michel, M.; Hutzler, M. Saccharomyces cerevisiae variety diastaticus friend or foe?—Spoilage potential and brewing ability of different Saccharomyces cerevisiae variety diastaticus yeast isolates by genetic, phenotypic and physiological characterization. FEMS Yeast Res. 2018, 18. [Google Scholar] [CrossRef]
- Gonçalves, M.; Pontes, A.; Almeida, P.; Barbosa, R.; Serra, M.; Libkind, D.; Hutzler, M.; Gonçalves, P.; Sampaio, J. Distinct domestication trajectories in top-fermenting beer yeasts and wine yeasts. Curr. Biol. 2016, 26, 2750–2761. [Google Scholar] [CrossRef] [Green Version]
- Fay, J.C.; Benavides, J.A. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 2005, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; Simpson, J.T.; Salinas, F.; Barré, B.P.; Parts, L.; Zia, A.; Ba, A.N.N.; Moses, A.M.; Louis, E.; Mustonen, V.; et al. A high-definition view of functional genetic variation from natural yeast genomes. Mol. Biol. Evol. 2014, 31, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Piškur, J.; Rozpedowska, E.; Polakova, S.; Merico, A.; Compagno, C. How did Saccharomyces evolve to become a good brewer? Trends Genet. 2006, 22, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Wiens, F.; Zitzmann, A.; Lachance, M.-A.; Yegles, M.; Pragst, F.; Wurst, F.M.; Von Holst, D.; Guan, S.L.; Spanagel, R. Chronic intake of fermented floral nectar by wild treeshrews. Proc. Natl. Acad. Sci. USA 2008, 105, 10426–10431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, F.; Aigle, M. Rtm1: A member of a new family of telomeric repeated genes in yeast. Genetics 1995, 140, 945–956. [Google Scholar]
- Borneman, A.R.; Pretorius, I.S. Genomic Insights into the Saccharomyces sensu stricto complex. Genetics 2015, 199, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.; Dietrich, F.S. The reacquisition of biotin prototrophy in Saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics 2007, 177, 2293–2307. [Google Scholar] [CrossRef] [Green Version]
- Naumov, G.; Turakainen, H.; Naumova, E.; Aho, S.; Korhola, M. A new family of polymorphic genes in Saccharomyces cerevisiae: α-galactosidase genes MEL1-MEL7. Mol. Genet. Genom. 1990, 224, 119–128. [Google Scholar] [CrossRef]
- Naumov, G.; Naumova, E.; Turakainen, H.; Suominen, P.; Korhola, M. Polymeric genes MEL8, MEL9 and MEL10 new members of α-galactosidase gene family in Saccharomyces cerevisiae. Curr. Genet. 1991, 20, 269–276. [Google Scholar] [CrossRef]
- Turakainen, H.; Aho, S.; Korhola, M. MEL gene polymorphism in the genus Saccharomyces. Appl. Environ. Microbiol. 1993, 59, 2622–2630. [Google Scholar] [CrossRef] [Green Version]
- Naumova, E.S.; Serpova, E.V.; Korshunova, I.V.; Naumov, G.I. Molecular polymorphism of α-galactosidase MEL genes of Saccharomyces yeasts. Microbiology 2011, 80, 502–513. [Google Scholar] [CrossRef]
- Naumov, G.I.; Naumova, E.S.; Korshunova, I.V.; Jakobsen, M. Yeast comparative genetics: a new MEL15 α-Galactosidase gene of Saccharomyces cerevisiae. Russ. J. Genet. 2002, 38, 1127–1132. [Google Scholar] [CrossRef]
- Naumova, E.S.; Korshunova, I.V.; Naumov, G.I. Molecular analysis of alpha-galactosidase MEL genes from Saccharomyces sensu stricto. Mol. Biol. 2003, 37, 699–703. [Google Scholar] [CrossRef]
- Fay, J.C.; Liu, P.; Ong, G.T.; Dunham, M.J.; Cromie, G.A.; Jeffery, E.W.; Ludlow, C.L.; Dudley, A.M. A polyploid admixed origin of beer yeasts derived from European and Asian wine populations. PLoS Biol. 2019, 17, e3000147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio, J.P.; Gonçalves, P. Biogeography and ecology of the genus Saccharomyces. In Yeasts in Natural Ecosystems: Ecology; Buzzini, P., Lachance, M.-A., Yurkov, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 131–153. [Google Scholar]
- Sampaio, J.; Gonçalves, P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl. Environ. Microbiol. 2008, 74, 2144–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, H.A.; Pinharanda, A.; Bensasson, D. Summer temperature can predict the distribution of wild yeast populations. Ecol. Evol. 2016, 6, 1236–1250. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Name | Strain Number# | Strain Number (Other Collections) | Strain Status | Population | Substrate of Isolation | Locality of Isolation |
|---|---|---|---|---|---|---|
| Saccharomyces aceti Santa María 1958 | CBS 4054 | – | type strain | WINE-PDM | red wine | Spain |
| Saccharomyces annulatus Negroni 1929 | PYCC 6727 | CBS 1227 | type strain | WINE-MAIN | abscess on epididymis | New Zealand |
| Saccharomyces boulardii Seguela, Bastide & Massot 1984 (invalid name) | YJS 5879 | - | type strain | WINE-MAIN | lychee | Vietnam |
| Saccharomyces brasiliensis Lindner 1909; Saccharomyces logos van Laer ex Jörgenssen 1909 | PYCC 4602 | CBS 382 | type strain | BEER 2- DIASTATICUS | beer (Logos brewery) | Rio de Janeiro, Brazil |
| Saccharomyces capensis van der Walt & Tscheuschner 1956 | CBS 2247 | - | type strain | WINE-PDM | grape must | South Africa |
| Saccharomyces cerevisiae Meyen ex Hansen 1883 | PYCC 4455 | CBS 1171 | neotype strain | BEER 1 | brewer’s top yeast | Oranjeboom brewery, Rotterdam, Netherlands |
| Saccharomyces cerevisiae Hansen var. onychophilus Zach 1934 | CBS 1464 | - | type strain | BREAD | nail of 4-year-old girl | Austria |
| Saccharomyces cerevisiae Hansen var. orsati Steiner 1924 | CBS 1175 | - | type strain | WINE-MAIN | wine | unknown |
| Saccharomyces chevalieri Guilliermond 1914 | PYCC 8489 | NRRL Y-12,633; CBS 400 | type strain | WEST AFRICA | palm wine from Elaeis guineensis | Ivory Coast |
| Saccharomyces chodati Steiner 1924 | CBS 423 | - | type strain | WINE-MAIN | wine | Switzerland |
| Saccharomyces cordubensis Santa María 1970 | CBS 6007 | - | type strain | WINE-PDM | wine | Spain |
| Saccharomyces coreanus Saito 1910 | CBS 5635 | - | neotype strain | WINE-MAIN | grape must | South Africa |
| Saccharomyces diastaticus Andrews & Gilliland ex van der Walt 1965 | PYCC 2608 | CBS 1782 | type strain | BEER 2-DIASTATICUS | super-attenuated beer | unknown |
| Saccharomyces carlsbergensis Hansen var. mandschuricus (Saito) Stelling-Dekker 1931 | CBS 1508 | - | type strain | SAKE | starter for sorghum brandy | unknown |
| Saccharomyces ellipsoideus Reess 1870 | PYCC 4653 | CBS 1395; NRRL Y-1529 | neotype strain | WINE—MAIN | unknown | unknown |
| Saccharomyces ellipsoideus Hansen var. umbra Castelli 1933 | CBS 457 | - | type strain | WINE—MAIN | grape must | Italy |
| Saccharomyces ellipsoideus Hansen var. alpinus Steiner 1924 | CBS 1192 | - | type strain | WINE—MAIN | wine | unknown |
| Saccharomyces ellipsoideus Hansen var. montibensis Steiner 1924 | CBS 1479 | - | type strain | WINE—MAIN | wine | unknown |
| Saccharomyces ellipsoideus Hansen ssp. thermophilus Steiner 1924 | CBS 1194 | - | type strain | WINE—MAIN | wine | unknown |
| Saccharomyces fructuum Lodder & Kreger-van Rij 1952 | PYCC 4654 | CBS 1544 | type strain | OUTLIER | fermenting fruit juice | Netherlands |
| Saccharomyces elongatus Krumbholz 1932 | CBS 439 | - | type strain | WINE—MAIN | Silvaner grapes | Germany |
| Saccharomyces formosensis Nakazawa 1933 | CBS 440 | - | type strain | SAKE | molasses | Taiwan |
| Saccharomyces gaditensis Santa María 1970 | CBS 6006 | - | type strain | WINE—PDM | wine | Spain |
| Saccharomyces hienipiensis Santa María 1962 | PYCC 6733 | VKM Y-1235 | type strain | OLIVES | alpechin | Spain |
| Saccharomyces hispalensis Santa María 1978 | PYCC 8023 | CBS 7002 | type strain | OLIVES | alpechin | Seville, Spain |
| Saccharomyces hispanica Santa María 1968 | CBS 5835 | - | type strain | WINE-PDM | wine | Spain |
| Saccharomyces ilicis Grönlund 1893 | CBS 1173 | - | type strain | OUTLIER | fruit of Ilex aquifolium | unknown |
| Saccharomyces italicus Castelli 1939 | CBS 459 | - | type strain | WINE—MAIN | grape must | Italy |
| Saccharomyces italicus var. melibiosi van Uden & Assis-Lopes 1957 | PYCC 2613 | CBS 2909 | type strain | OLIVES | feces of man | Portugal |
| Saccharomyces lindneri Guilliermond 1914 | CBS 403 | PYCC 4571 | type strain | WEST AFRICA | ginger beer from Zinziber officinale | West Africa |
| Saccharomyces mangini var. casei Saccchetti 1933 | PYCC 6721 | CBS 420; VKM Y-482 | type strain | DAIRY | stracchino cheese | Italy |
| Saccharomyces marchalianus Kufferath 1920 | PYCC 8196 | CBS 1460 | type strain | WINE—MAIN | fermenting fruit | Indonesia |
| Saccharomyces multisporus Jörgensen 1909 | CBS 4507 | - | type strain | WINE—MAIN | English top brewing yeast | unknown |
| Population | Designation in Other Studies | Life Style (W-Wild; D-Domesticated; F-Feral) | Number of Type Strains of Former Saccharomyces Species | Comments |
|---|---|---|---|---|
| 1–Wine-main | Wine [25]; Wine/European [6,9] | D | 17 | Global distribution probably associated with the widespread dissemination of winemaking |
| 2–Olives | Alpechin [9]; Olives [26,27] | D | 6 | Ancestral hybrids with S. paradoxus associated with processed olives |
| 3–Wine-PDM | Flor [25,28]; Georgian [9]; PDM-Prise de Mousse [12]; | D | 6 | Lalvin EC1118, a well-known commercial strain in this group is also known as “Prise de mousse” or Champagne strain [20,28]. This population also contains the Spanish flor yeasts of Xerez wine (Sherry) and similar wines [28] |
| 4–Cachaça-Bioethanol | Brazilian bioethanol [9]; Cachaça C1 and C2 [29]; Rum and bioethanol [25] | D | – | This group is a secondary domesticate derived from wine strains and includes two types of cachaça strains (C1 and C2) and bioethanol strains [29] |
| 5–Beer 2-Diastaticus | Beer 2 [30]; Mosaic beer [9] | D | 6 | Contains Saison-type low-gravity beer strains and beer-spoilage strains with diastase activity |
| 6–African beer | African beer [9] | D | – | Includes strains that ferment malted millet to produce bantu beer or malted sorghum to produce bili-bili or kaffir beer |
| 7–Wild Brazil 3-French Guiana–Mexican agave | Mexican agave, French Guiana human [9]; Wild Brazil B3 [31] | W/D | – | Complex clade composed of three subclades, one containing wild strains found in Brazil, another containing strains used in artisanal mezcal fermentation in Mexico and a third one containing gut-associated strains from French Guiana aborigines; this last group contains STA1 and was also found in cachiri, a traditional beer made from chewed and fermented starch-rich manioc [32] |
| 8–Mediterranean oak | - | W | – | Unique wild population with no evident links to the core group of Asian wild populations; no changes in designation since the original description [8] |
| 9–Dairy | Cheese [25]; French dairy [9]; Milk [33] | D | 1 | Recently revealed population associated with dairy products and adapted to galactose utilization |
| 10–Bread | Active dry yeast [33]; Mixed [30] | D | 2 | |
| 11–Beer 1 | Ale beer [9]; Beer [25]; Beer 1 [30] | D | 1 | Various sub-populations associated with different ale-beer types |
| 12–Chinese fermentations (bread and beverages) | Mantou (bread)/Baijiu (distilled)/Huangjiu (rice wine)/Qingkejiu (barley wine)/fermented milk [33] | D | – | Predominantly Chinese domesticated strains that are distinct from strains of the Sake clade |
| 13–Cocoa | West African cocoa [9] | D | – | |
| 14–China VIII | - | F | – | Found in the arboreal niche in China, but with several domestication signatures (MAL gene expansion, AQY inactivation, presence of region B) |
| 15–Sake | Asia [27,30]; Asian fermentation, Sake [6,9] | D | 3 | |
| 16–Philippines | Asian islands [9]; Philippines [27] | W/D | – | |
| 17–North America–Japan-Far East Russia–China VI-VII | China VI-VII [33,34]; Far East Russia [9]; North American [6] | W | – | A wild Chinese population also found in North America, Japan and Russia |
| 18–China VI-VII | - | W | – | No changes in designation since the original description [34] |
| 19–West Africa | African palm wine [9]; West Africa [6]; | D | 2 | |
| 20–Wild Brazil 1–China V | Ecuadorean [9]; Wild Brazil B1 [31] | W | – | A wild Chinese population also found in Brazil and Ecuador |
| 21–China I | - | W | – | No changes in designation since the original description [34] |
| 22–Malaysia | - | W | – | No changes in designation since the original description [6] |
| 23–China III | - | W | – | No changes in designation since the original description [34] |
| 24–China IV | - | W | – | No changes in designation since the original description [34] |
| 25–China I | - | W | – | No changes in designation since the original description [34] |
| 26–China IX | - | W | – | No changes in designation since the original description [34] |
| 27–China II | - | W | – | No changes in designation since the original description [34] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontes, A.; Hutzler, M.; Brito, P.H.; Sampaio, J.P. Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication. Microorganisms 2020, 8, 903. https://doi.org/10.3390/microorganisms8060903
Pontes A, Hutzler M, Brito PH, Sampaio JP. Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication. Microorganisms. 2020; 8(6):903. https://doi.org/10.3390/microorganisms8060903
Chicago/Turabian StylePontes, Ana, Mathias Hutzler, Patrícia H. Brito, and José Paulo Sampaio. 2020. "Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication" Microorganisms 8, no. 6: 903. https://doi.org/10.3390/microorganisms8060903
APA StylePontes, A., Hutzler, M., Brito, P. H., & Sampaio, J. P. (2020). Revisiting the Taxonomic Synonyms and Populations of Saccharomyces cerevisiae—Phylogeny, Phenotypes, Ecology and Domestication. Microorganisms, 8(6), 903. https://doi.org/10.3390/microorganisms8060903