Population Analysis and Evolution of Saccharomyces cerevisiae Mitogenomes


 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Collection
2.2. Alignment
2.3. General Statistics and Linkage Disequilibrium
2.4. Genetic Structure of S. cerevisiae mtDNA
3. Results
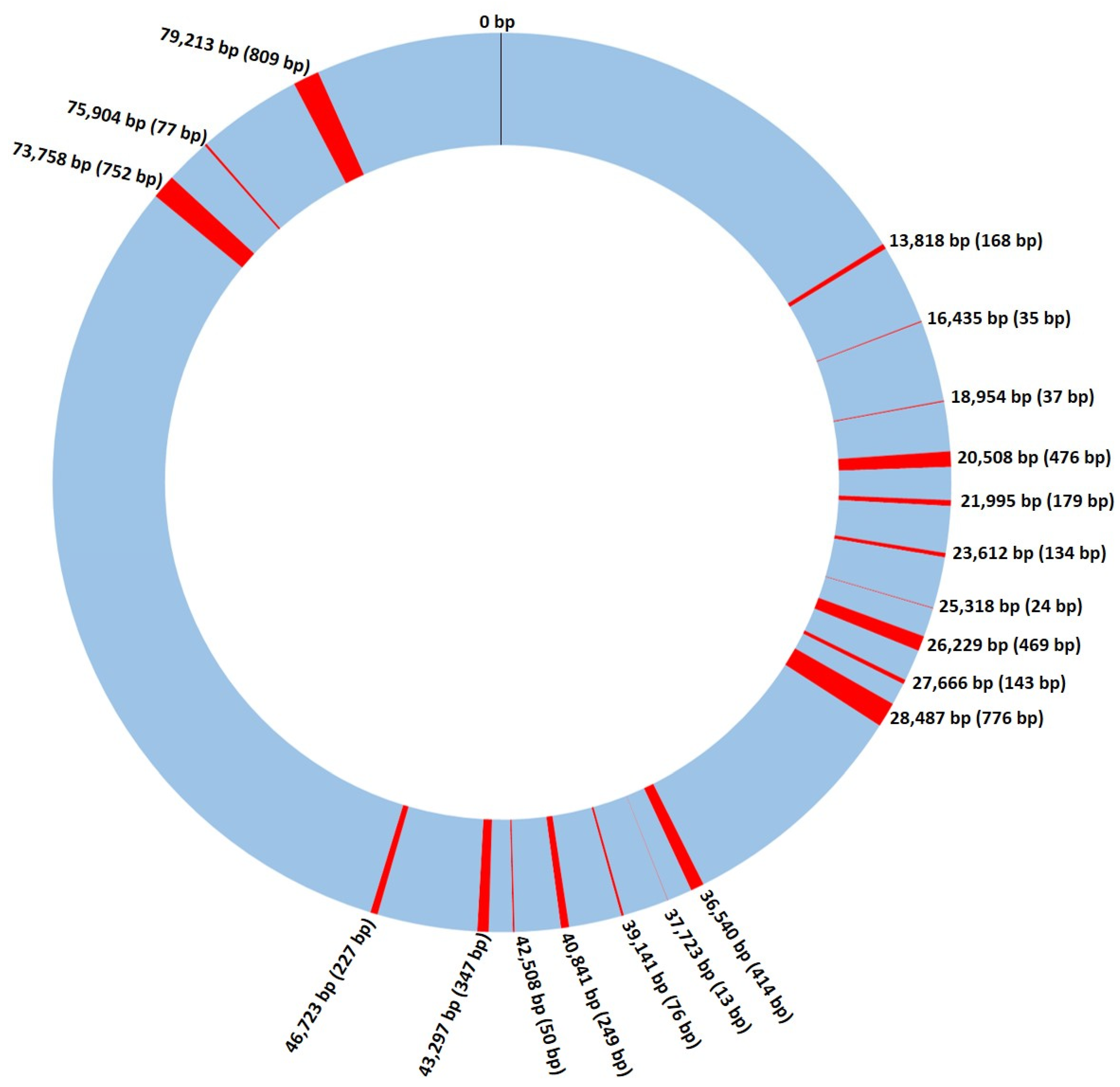
3.1. Saccharomyces cerevisiae Mitochondrial Genome
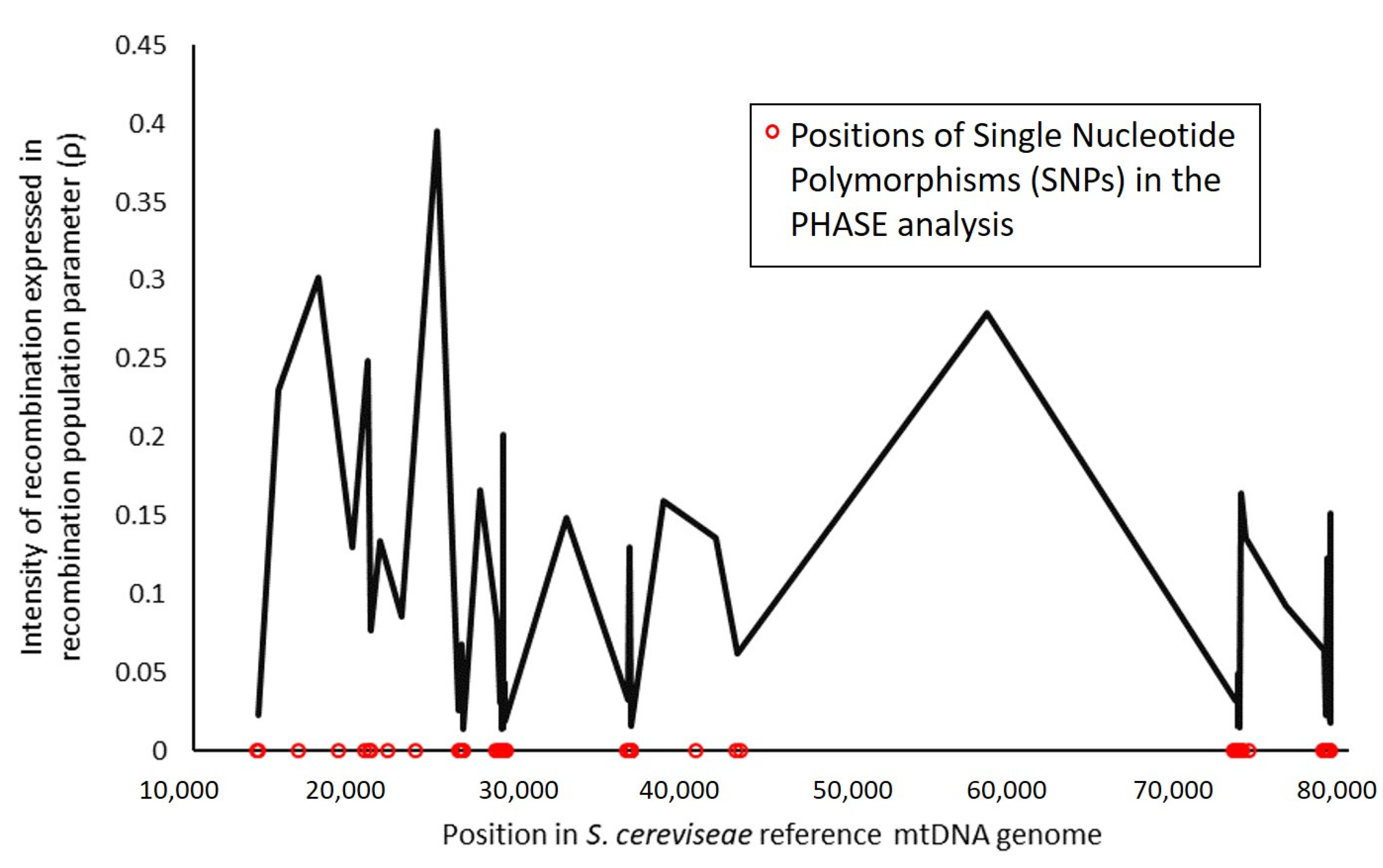
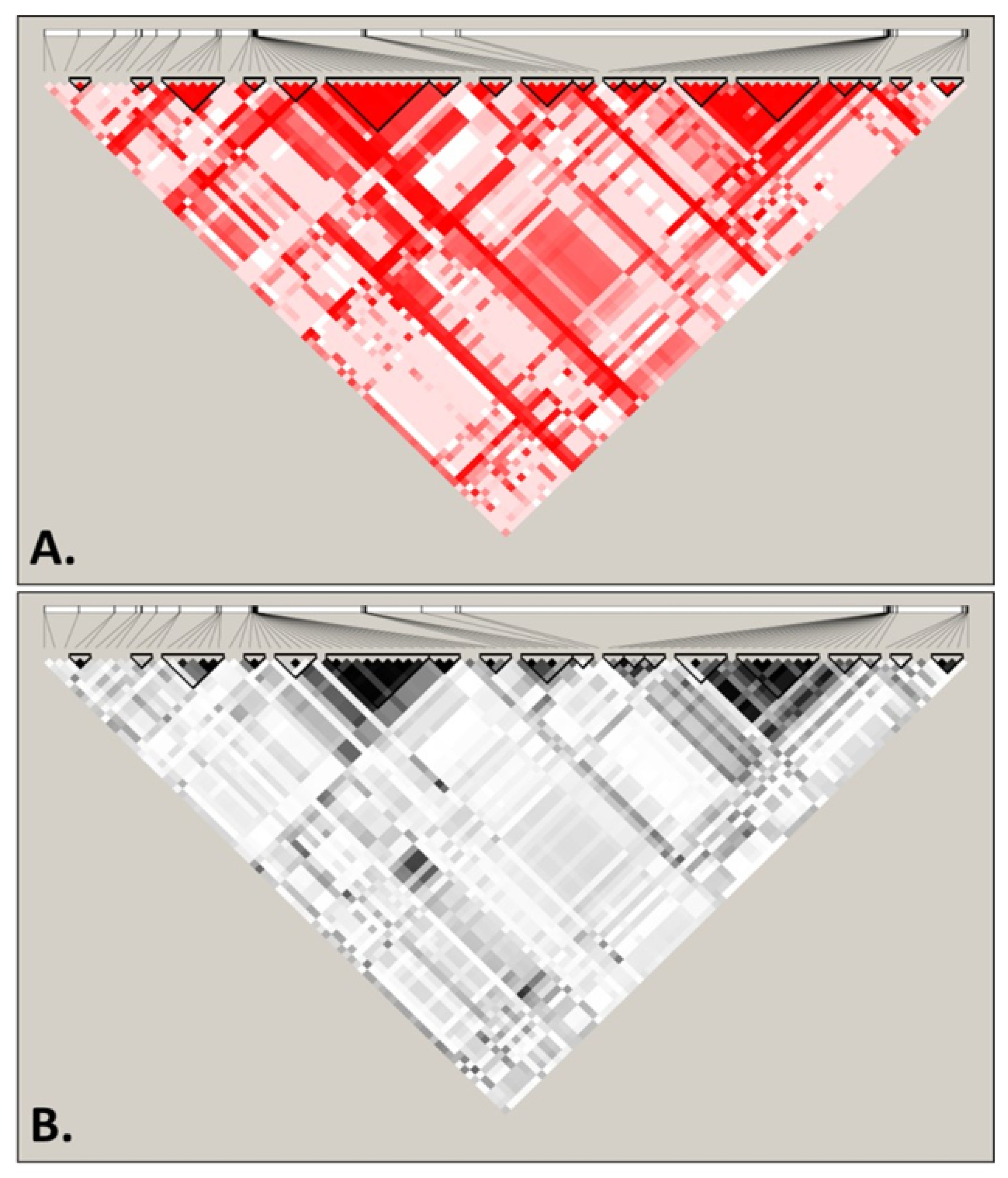
3.2. Recombination and Linkage Disequilibrium in S. cerevisiae Mitochondrial DNA
3.3. Genetic Structure of S. cerevisiae Mitochondrial DNA
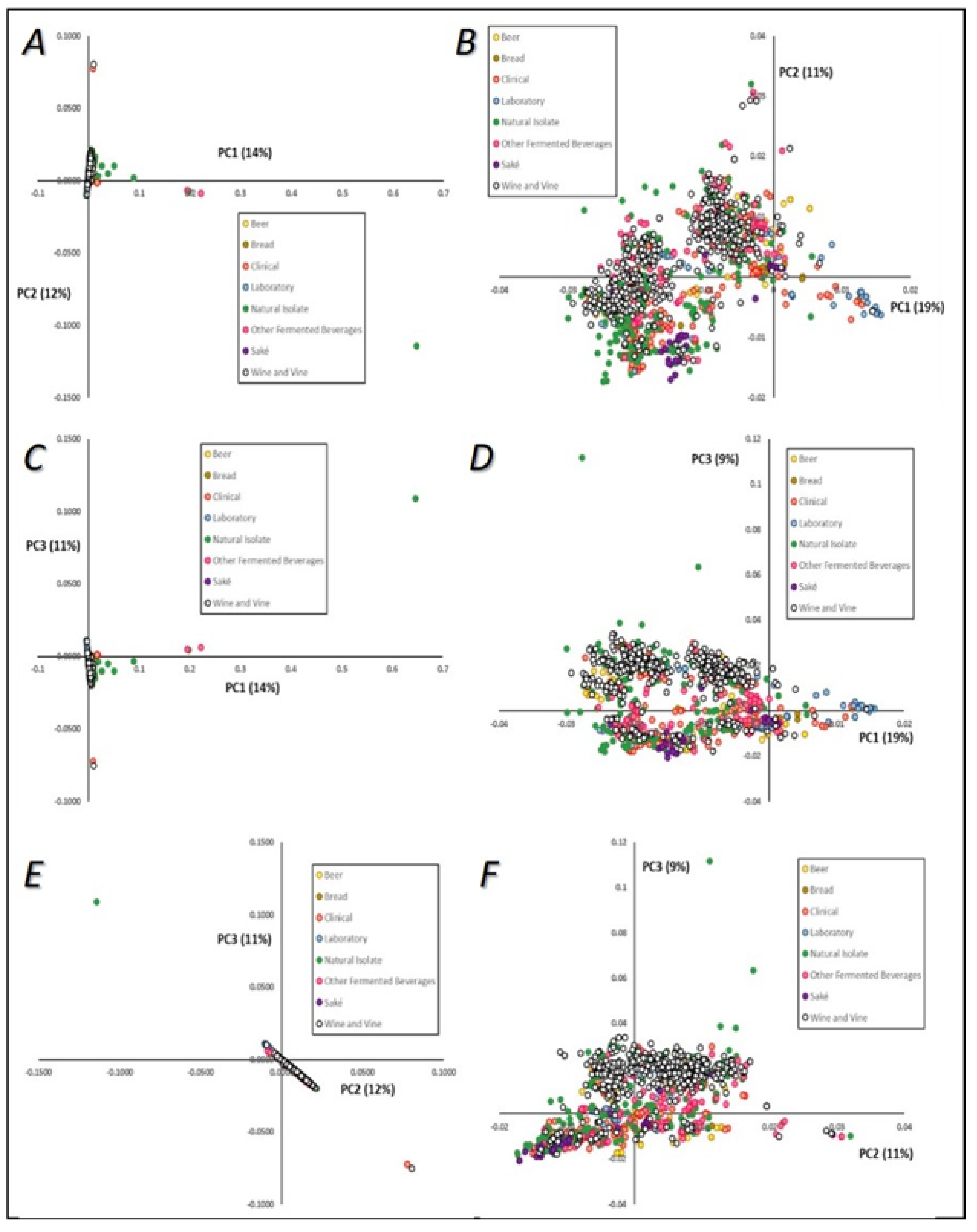
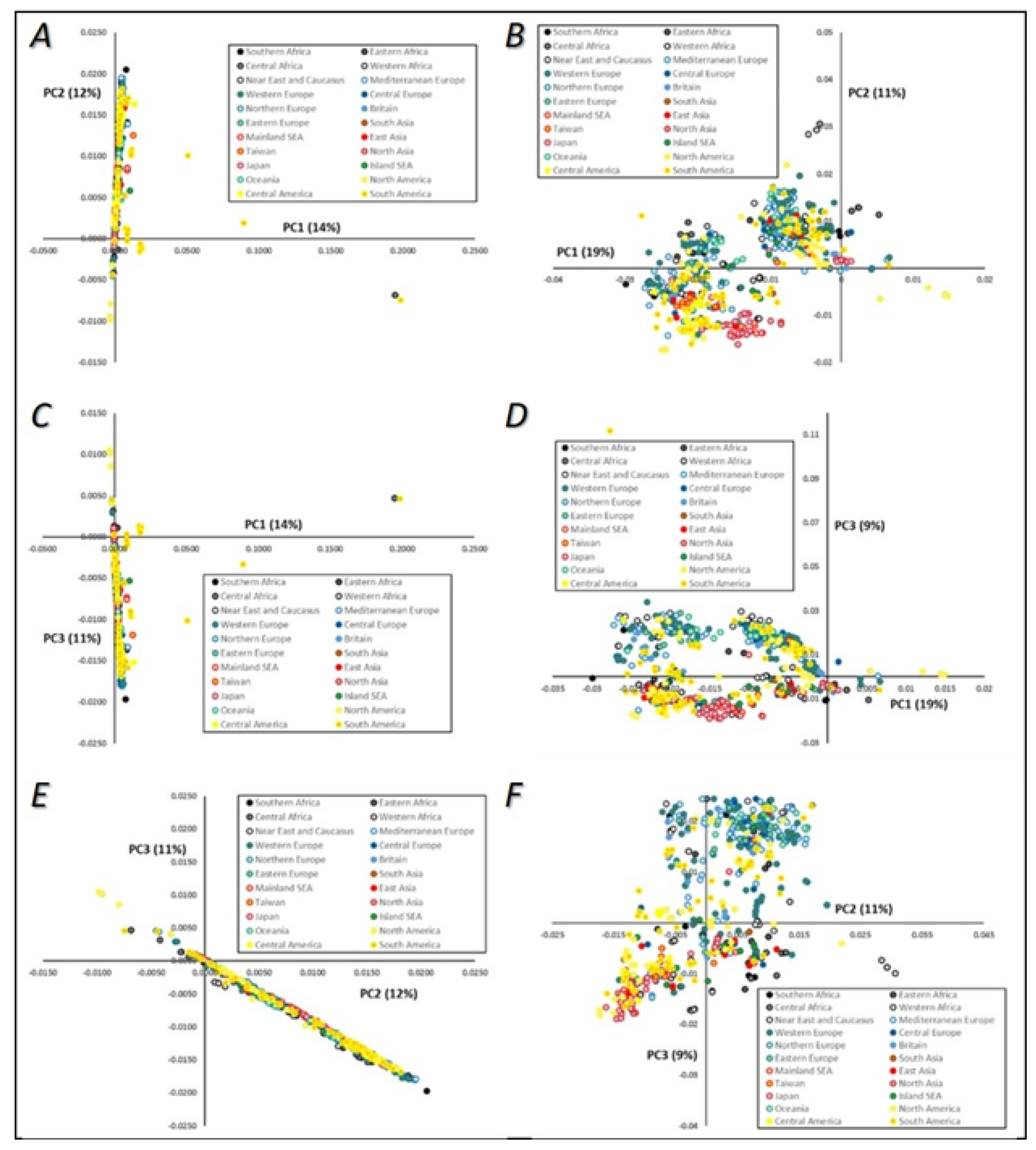
3.3.1. Principal Component Analysis (PCA)
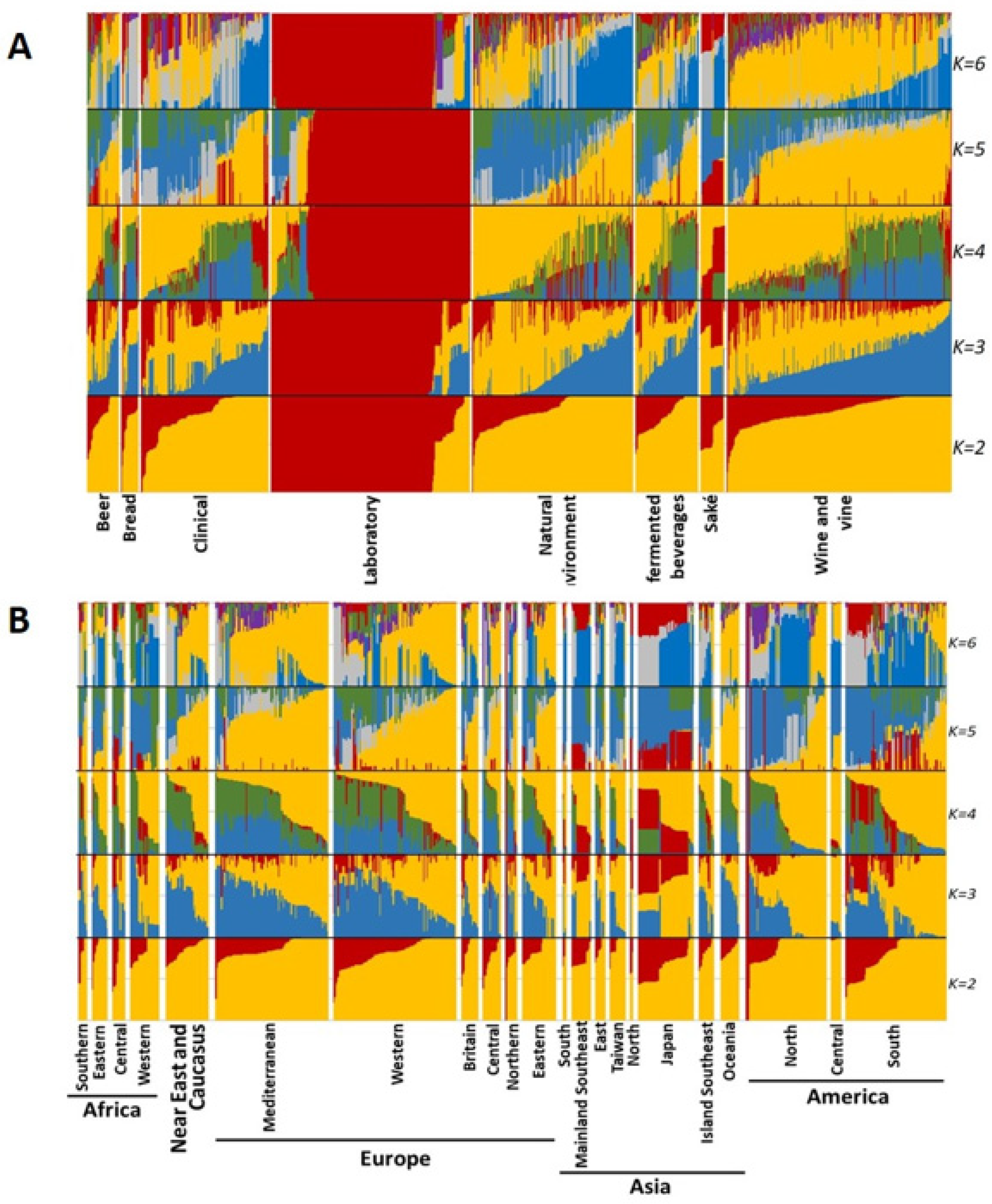
3.3.2. Sparse Non-Negative Matrix Factorization (sNMF)
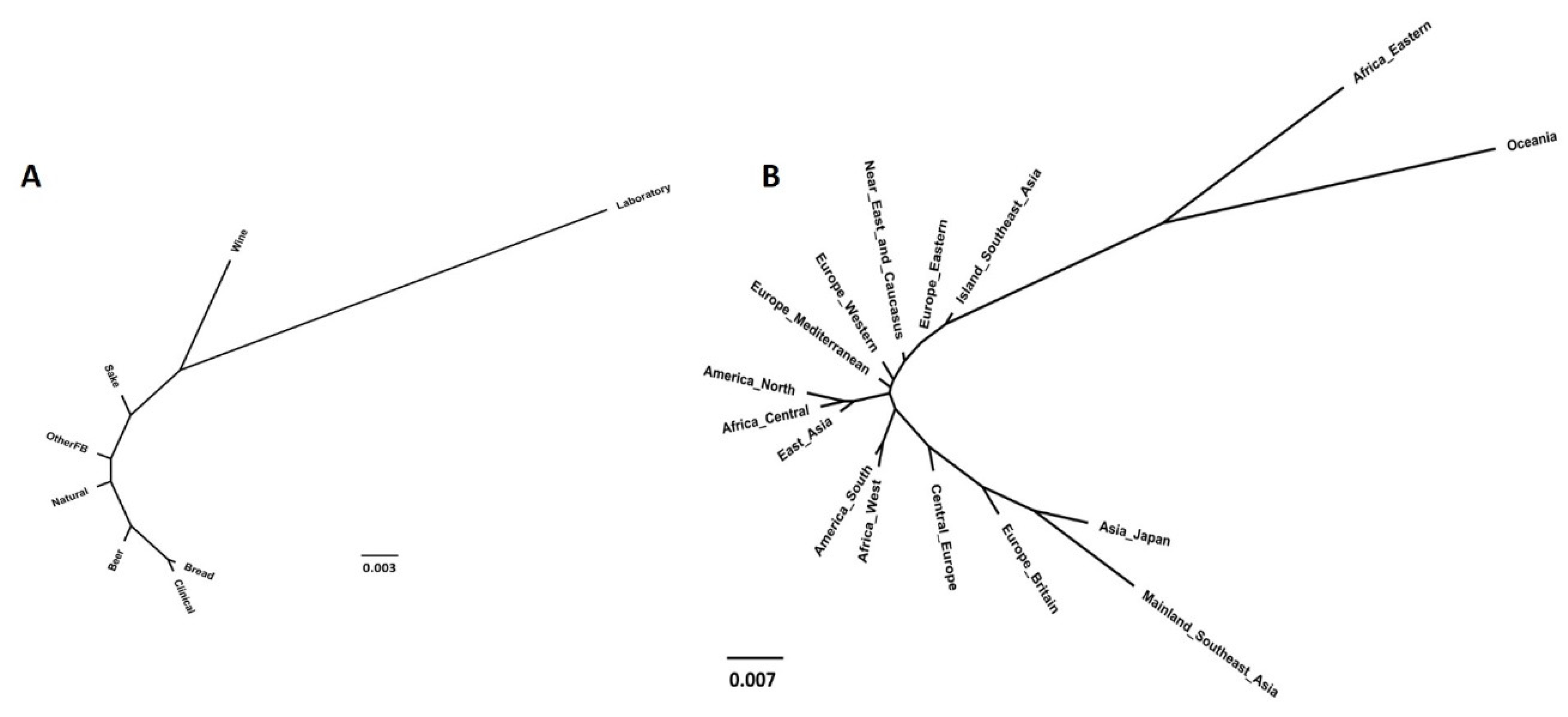
3.3.3. Neighbor-Joining Phylogenetic Trees
3.4. Population Structure Based on Strains’ Technological Sources and Geographical Origins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liti, G.; Carter, D.M.; Moses, A.M.; Warringer, J.; Parts, L.; James, S.A.; Davey, R.P.; Roberts, I.N.; Burt, A.; Koufopanou, V.; et al. Population genomics of domestic and wild yeasts. Nature 2009, 458, 337–341. [Google Scholar] [CrossRef]
- De Deken, R.H. The crabtree effect: A regulatory system in yeast. J. Gen. Microbiol. 1966, 44, 149–156. [Google Scholar] [CrossRef]
- Gancedo, J.M. Yeast carbon catabolite repression. Microbiol. Mol. Biol. Rev. 1998, 62, 28. [Google Scholar] [CrossRef]
- Piskur, J.; Rozpedowska, E.; Polakova, S.; Merico, A.; Compagno, C. How did Saccharomyces evolve to become a good brewer? Trends Genet. 2006, 22, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces genome database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M. The yeast genome: On the road to the golden age. Curr. Opin. Genet. Dev. 2000, 10, 617–623. [Google Scholar] [CrossRef]
- Wang, Q.-M.; Liu, W.-Q.; Liti, G.; Wang, S.-A.; Bai, F.-Y. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol. Ecol. 2012, 21, 5404–5417. [Google Scholar] [CrossRef]
- Matti, S. Oxidative Phosphorylation at the fin de siècle. Science 1999, 283, 1488–1493. [Google Scholar]
- Hsu, Y.-Y.; Chou, J.-Y. Environmental factors can influence mitochondrial inheritance in the Saccharomyces yeast hybrids. PLoS ONE 2017, 12, e0169953. [Google Scholar] [CrossRef]
- Wallace, D.C. Why do we still have a maternally inherited mitochondrial dna? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. 2012, 4, a011403. [Google Scholar] [CrossRef]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. PNAS 2009, 106, 11960–11965. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Solieri, L. Mitochondrial inheritance in budding yeasts: Towards an integrated understanding. Trends Microbiol. 2010, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.P.; Friedrich, A.; Reisser, C.; Hou, J.; Schacherer, J. Mitochondrial genome evolution in a single protoploid yeast species. G3 2012, 2, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Lu, K.; Reichert, A.S. Mitophagy and mitochondrial dynamics in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2015, 1853, 2766–2774. [Google Scholar] [CrossRef]
- Preuten, T.; Cincu, E.; Fuchs, J.; Zoschke, R.; Liere, K.; Börner, T. Fewer genes than organelles: Extremely low and variable gene copy numbers in mitochondria of somatic plant cells: Gene copy numbers in mitochondria. Plant J. 2010, 64, 948–959. [Google Scholar] [CrossRef]
- Shay, W.; Piercel, J. Mitochondrial DNA copy number is proportional to total cell DNA under a variety of growth conditions. J. Biol. Chem. 1990, 265, 14802–14807. [Google Scholar]
- Hori, A.; Yoshida, M.; Shibata, T.; Ling, F. Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination-mediated replication. Nucleic Acids Res. 2009, 37, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, I. Life Cycle of the Budding Yeast Saccharomyces cerevisiae. Microbiol. Rev. 1988, 52, 18. [Google Scholar] [CrossRef]
- Hittinger, C.T. Saccharomyces diversity and evolution: A budding model genus. Trends Genet. 2013, 29, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wolters, J.F.; Charron, G.; Gaspary, A.; Landry, C.R.; Fiumera, A.C.; Fiumera, H.L. Mitochondrial recombination reveals mito–mito epistasis in yeast. Genetics 2018, 209, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Verspohl, A.; Pignedoli, S.; Giudici, P. The inheritance of mitochondrial DNA in interspecific Saccharomyces hybrids and their properties in winemaking: Mitochondrial DNA inheritance in interspecific Saccharomyces hybrids. Yeast 2018, 35, 173–187. [Google Scholar] [CrossRef]
- Lipinski, K.A.; Kaniak-Golik, A.; Golik, P. Maintenance and expression of the S. cerevisiae mitochondrial genome—From genetics to evolution and systems biology. Biochim. Biophys. Acta 2010, 1797, 1086–1098. [Google Scholar] [CrossRef]
- Goddard, M.R.; Burt, A. Recurrent invasion and extinction of a selfish gene. PNAS 1999, 96, 13880–13885. [Google Scholar] [CrossRef]
- Jakobs, S. Spatial and temporal dynamics of budding yeast mitochondria lacking the division component Fis1p. J. Cell Sci. 2003, 116, 2005–2014. [Google Scholar] [CrossRef]
- Merz, S.; Hammermeister, M.; Altmann, K.; Dürr, M.; Westermann, B. Molecular machinery of mitochondrial dynamics in yeast. Biol. Chem. 2007, 388, 917–926. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial dynamics in model organisms: What yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin. Cell Dev. Biol. 2010, 21, 542–549. [Google Scholar] [CrossRef]
- Simon, V.R.; Karmon, S.L.; Pon, L.A. Mitochondrial inheritance: Cell cycle and actin cable dependence of polarized mitochondrial movements in Saccharomyces cerevisiae. Cell Motil. Cytoskelet. 1997, 37, 199–210. [Google Scholar] [CrossRef]
- Yang, H.-C.; Palazzo, A.; Swayne, T.C.; Pon, L.A. A retention mechanism for distribution of mitochondria during cell division in budding yeast. Curr. Biol. 1999, 9, 1111–1114. [Google Scholar] [CrossRef]
- Fehrenbacher, K.L.; Yang, H.-C.; Gay, A.C.; Huckaba, T.M.; Pon, L.A. Live cell imaging of mitochondrial movement along actin cables in budding yeast. Curr. Biol. 2004, 14, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial inheritance in yeast. Biochim. Et Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Turk, E.M.; Das, V.; Seibert, R.D.; Andrulis, E.D. The Mitochondrial RNA landscape of Saccharomyces cerevisiae. PLoS ONE 2013, 8, e78105. [Google Scholar] [CrossRef]
- Schacherer, J.; Shapiro, J.A.; Ruderfer, D.M.; Kruglyak, L. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 2009, 458, 342–345. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 2019, 15. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.G. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Direct evidence for homologous recombination in mussel (Mytilus galloprovincialis) mitochondrial DNA. Mol. Biol. Evol. 2001, 18, 1168–1175. [Google Scholar] [CrossRef]
- Leducq, J.-B.; Henault, M.; Charron, G.; Nielly-Thibault, L.; Terrat, Y.; Fiumera, H.L.; Shapiro, B.J.; Landry, C.R. Mitochondrial recombination and introgression during speciation by hybridization. Mol. Biol. Evol. 2017, 34, 1947–1959. [Google Scholar] [CrossRef]
- Fritsch, E.S.; Chabbert, C.D.; Klaus, B.; Steinmetz, L.M. A genome-wide map of mitochondrial DNA recombination in yeast. Genetics 2014, 198, 755–771. [Google Scholar] [CrossRef] [PubMed]
- Warringer, J.; Zörgö, E.; Cubillos, F.A.; Zia, A.; Gjuvsland, A.; Simpson, J.T.; Forsmark, A.; Durbin, R.; Omholt, S.W.; Louis, E.J.; et al. Trait variation in yeast is defined by population history. PLoS Genet. 2011, 7, e1002111. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, L.N.; Brem, R.B.; Kruglyak, L.; Gottschling, D.E. Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics 2009, 183, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The petite mutation in yeast. Trends Biochem. Sci. 1979, 4, 197–201. [Google Scholar] [CrossRef]
- Dujon, B.; Slonimski, P.P. Mitochondrial Genetics IX: A model for recombination and segregation of mitochondrial genomes in Saccharomyces cerevisiae. Genetics 1974, 78, 415–437. [Google Scholar] [PubMed]
- Piškur, J. Transmission of yeast mitochondrial loci to progeny is reduced when nearby intergenic regions containing ori sequences are deleted. Mol. Gen. Genet. 1988, 214, 425–432. [Google Scholar] [CrossRef]
- Clark-Walker, G.D. In vivo rearrangement of mitochondrial DNA in Saccharomyces cerevisiae. PNAS 1989, 86, 8847–8851. [Google Scholar] [CrossRef]
- Sulo, P.; Szabóová, D.; Bielik, P.; Poláková, S.; Šoltys, K.; Jatzová, K.; Szemes, T. The evolutionary history of Saccharomyces species inferred from completed mitochondrial genomes and revision in the ‘yeast mitochondrial genetic code’. DNA Res. 2017, 24, 571–583. [Google Scholar] [CrossRef]
- Zinn, A.R.; Pohlman, J.K.; Perlman, P.S.; Butow, R.A. Kinetic and segregational analysis of mitochondrial DNA recombination in yeast. Plasmid 1987, 17, 248–256. [Google Scholar] [CrossRef]
- Perez-Martinez, X. Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 2003, 22, 5951–5961. [Google Scholar] [CrossRef]
- Wu, B.; Hao, W. Mitochondrial-encoded endonucleases drive recombination of protein-coding genes in yeast. Environ. Microbiol. 2019, 21, 4233–4240. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Jobin, M.; Schurz, H.; Henn, B.M. IMPUTOR: Phylogenetically aware software for imputation of errors in next-generation sequencing. Genome Biol. Evol. 2018, 10, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 117693430500100. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Crawford, D.C.; Bhangale, T.; Li, N.; Hellenthal, G.; Rieder, M.J.; Nickerson, D.A.; Stephens, M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 2004, 36, 700–706. [Google Scholar] [CrossRef]
- Li, N.; Stephens, M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 2003, 165, 2213–2233. [Google Scholar]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; Mathieu, F.; Trouillon, T.; Bouchard, G.; François, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 2014, 196, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Abrantes, D.; Rito, T.; Thomson, N.; Radivojac, P.; Li, B.; Macaulay, V.; Samuels, D.C.; Pereira, L. Evaluating purifying selection in the mitochondrial DNA of various mammalian species. PLoS ONE 2013, 8, e58993. [Google Scholar] [CrossRef] [PubMed]
- Mendes, I.; Franco-Duarte, R.; Umek, L.; Fonseca, E.; Drumonde-Neves, J.; Dequin, S.; Zupan, B.; Schuller, D. Computational Models for prediction of yeast strain potential for winemaking from phenotypic profiles. PLoS ONE 2013, 8, e66523. [Google Scholar] [CrossRef]
- Franco-Duarte, R.; Mendes, I.; Umek, L.; Drumonde-Neves, J.; Zupan, B.; Schuller, D. Computational models reveal genotype-phenotype associations in Saccharomyces cerevisiae: Genetic and phenotypic relationships in a strain collection. Yeast 2014, 31, 265–277. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 4th ed.; Sinauer Associates Inc. Publishers: Sunderland, MA, USA, 1997. [Google Scholar]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef]
- Freel, K.C.; Friedrich, A.; Schacherer, J. Mitochondrial genome evolution in yeasts: An all-encompassing view. FEMS Yeast Res. 2015, 15, fov023. [Google Scholar] [CrossRef]
- Casaregola, S.; Nguyen, H.V.; Lepingle, A.; Brignon, P.; Gendre, F.; Gaillardin, C. A family of laboratory strains of Saccharomyces cerevisiae carry rearrangements involving chromosomes I and III. Yeast 1998, 14, 551–564. [Google Scholar] [CrossRef]
- Daranlapujade, P.; Daran, J.; Kotter, P.; Petit, T.; Piper, M.; Pronk, J. Comparative genotyping of the laboratory strains S288C and CEN.PK113-7D using oligonucleotide microarrays. Fems. Yeast Res. 2003, 4, 259–269. [Google Scholar] [CrossRef][Green Version]
- Pizarro, F.J.; Jewett, M.C.; Nielsen, J.; Agosin, E. Growth temperature exerts differential physiological and transcriptional responses in laboratory and wine strains of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2008, 74, 6358–6368. [Google Scholar] [CrossRef]
- Franco-Duarte, R.; Umek, L.; Mendes, I.; Castro, C.C.; Fonseca, N.; Martins, R.; Silva-Ferreira, A.C.; Sampaio, P.; Pais, C.; Schuller, D. New integrative computational approaches unveil the Saccharomyces cerevisiae pheno-metabolomic fermentative profile and allow strain selection for winemaking. Food Chem. 2016, 211, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Mendes, I.; Sanchez, I.; Franco-Duarte, R.; Camarasa, C.; Schuller, D.; Dequin, S.; Sousa, M.J. Integrating transcriptomics and metabolomics for the analysis of the aroma profiles of Saccharomyces cerevisiae strains from diverse origins. BMC Genom. 2017, 18, 455. [Google Scholar] [CrossRef] [PubMed]
- Legras, J.-L.; Galeote, V.; Bigey, F.; Camarasa, C.; Marsit, S.; Nidelet, T.; Sanchez, I.; Couloux, A.; Guy, J.; Franco-Duarte, R.; et al. Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Mol. Biol. Evol. 2018, 35, 1712–1727. [Google Scholar] [CrossRef] [PubMed]
- Franco-Duarte, R.; Bessa, D.; Gonçalves, F.; Martins, R.; Silva-Ferreira, A.C.; Schuller, D.; Sampaio, P.; Pais, C. Genomic and transcriptomic analysis of Saccharomyces cerevisiae isolates with focus in succinic acid production. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Franco-Duarte, R.; Bigey, F.; Carreto, L.; Mendes, I.; Dequin, S.; Santos, M.A.; Pais, C.; Schuller, D. Intrastrain genomic and phenotypic variability of the commercial Saccharomyces cerevisiae strain Zymaflore VL1 reveals microevolutionary adaptation to vineyard environments. FEMS Yeast Res. 2015, 15, fov063. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fontenot, B.E.; Makowsky, R.; Chippindale, P.T. Nuclear–mitochondrial discordance and gene flow in a recent radiation of toads. Mol. Phylogenetics Evol. 2011, 59, 66–80. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Parameter | Statistics |
|---|---|
| Number of sequences | 6646 |
| Size (bp) | 5475 |
| Number of polymorphic sites | 526 |
| Number of haplotypes | 1265 |
| Number of observed transitions | 273 |
| Number of observed transversion | 291 |
| Nucleotide composition (%)—C | 13.09 |
| Nucleotide composition (%)—T | 41.33 |
| Nucleotide composition (%)—A | 30.95 |
| Nucleotide composition (%)—G | 14.63 |
| Gene diversity | 0.9665 ± 0.0032 |
| Mean number of pairwise differences | 38.632869 ± 16.797110 |
| Nucleotide diversity (average over loci) | 0.007354 ± 0.003535 |
| Tajima’s D p-value | 0.10600 |
| Fu’s FS p-value | 0.59400 |
| Beer | Bread | Clinical | Laboratory | Natural | Other Fb | Saké | Wine and Vine | |
|---|---|---|---|---|---|---|---|---|
| - | 0.0031 | 0.0034 | 0.0543 | 0.0031 | 0.0009 | 0.0037 | 0.0025 | Beer |
| - | 0.0016 | 0.0545 | 0.0075 | 0.0127 | 0.0097 | 0.0025 | Bread | |
| - | 0.0666 | 0.0035 | 0.0073 | 0.0133 | 0.0056 | Clinical | ||
| - | 0.0374 | 0.0312 | 0.0281 | 0.0235 | Laboratory | |||
| - | 0.0003 | 0.0024 | 0.0028 | Natural | ||||
| - | 0.0028 | 0.0038 | Other Fb | |||||
| - | 0.0020 | Saké | ||||||
| - | Wine and vine |
| Africa_Central | Africa_Eastern | Africa_West | America_North | America_South | East_Asia | Island_Southeast_Asia | Asia_Japan | Mainland_Southeast_Asia | Europe_Britain | Central_Europe | Europe_Eastern | Europe_Mediterranean | Europe_Western | Near_East_and_Caucasus | Oceania | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - | 0.0238 | 0.0127 | 0.0079 | 0.0054 | 0.0054 | 0.0222 | 0.0033 | 0.0268 | 0.0459 | 0.0215 | 0.0083 | 0.0152 | 0.0181 | 0.0162 | 0.0769 | Africa_Central |
| - | 0.0263 | 0.0078 | 0.0002 | 0.0342 | 0.0316 | 0.0184 | 0.0484 | 0.0229 | 0.0179 | 0.0299 | 0.0185 | 0.0184 | 0.0295 | 0.0154 | Africa_Eastern | |
| - | 0.0162 | 0.0012 | 0.0164 | 0.0120 | 0.0046 | 0.0322 | 0.0209 | 0.0245 | 0.0100 | 0.0150 | 0.0115 | 0.0170 | 0.0888 | Africa_West | ||
| - | 0.0107 | 0.0091 | 0.0202 | 0.0452 | 0.0733 | 0.0018 | 0.0137 | 0.0252 | 0.0198 | 0.0186 | 0.0116 | 0.0904 | America_North | |||
| - | 0.0149 | 0.0203 | 0.0067 | 0.0303 | 0.0129 | 0.0118 | 0.0220 | 0.0026 | 0.0069 | 0.0126 | 0.1132 | America_South | ||||
| - | 0.0195 | 0.0383 | 0.0779 | 0.0031 | 0.0197 | 0.0211 | 0.0028 | 0.0073 | 0.0163 | 0.0460 | East_Asia | |||||
| - | 0.0006 | 0.0247 | 0.0352 | 0.0224 | 0.0104 | 0.0177 | 0.0172 | 0.0109 | 0.0459 | Island_Souhteast_Asia | ||||||
| - | 0.0082 | 0.0026 | 0.0185 | 0.0239 | 0.0098 | 0.0121 | 0.0344 | 0.1680 | Asia_Japan | |||||||
| - | 0.0123 | 0.0071 | 0.0391 | 0.0339 | 0.0390 | 0.0638 | 0.1946 | Mainland_Southeast_Asia | ||||||||
| - | 0.0164 | 0.0003 | 0.0218 | 0.0189 | 0.0175 | 0.0921 | Europe_Britain | |||||||||
| - | 0.0167 | 0.0156 | 0.0146 | 0.0033 | 0.1049 | Central_Europe | ||||||||||
| - | 0.0031 | 0.0031 | 0.0060 | 0.0424 | Europe_Eastern | |||||||||||
| - | 0.0056 | 0.0031 | 0.1090 | Europe_Mediterranean | ||||||||||||
| - | 0.0062 | 0.0892 | Europe_Western | |||||||||||||
| - | 0.0490 | Near_East_and_Caucasus | ||||||||||||||
| - | Oceania |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, D.; Esteves, S.; Santiago, C.; Conde-Sousa, E.; Fernandes, T.; Pais, C.; Soares, P.; Franco-Duarte, R. Population Analysis and Evolution of Saccharomyces cerevisiae Mitogenomes. Microorganisms 2020, 8, 1001. https://doi.org/10.3390/microorganisms8071001
Vieira D, Esteves S, Santiago C, Conde-Sousa E, Fernandes T, Pais C, Soares P, Franco-Duarte R. Population Analysis and Evolution of Saccharomyces cerevisiae Mitogenomes. Microorganisms. 2020; 8(7):1001. https://doi.org/10.3390/microorganisms8071001
Chicago/Turabian StyleVieira, Daniel, Soraia Esteves, Carolina Santiago, Eduardo Conde-Sousa, Ticiana Fernandes, Célia Pais, Pedro Soares, and Ricardo Franco-Duarte. 2020. "Population Analysis and Evolution of Saccharomyces cerevisiae Mitogenomes" Microorganisms 8, no. 7: 1001. https://doi.org/10.3390/microorganisms8071001
APA StyleVieira, D., Esteves, S., Santiago, C., Conde-Sousa, E., Fernandes, T., Pais, C., Soares, P., & Franco-Duarte, R. (2020). Population Analysis and Evolution of Saccharomyces cerevisiae Mitogenomes. Microorganisms, 8(7), 1001. https://doi.org/10.3390/microorganisms8071001