Comparative Genomics of Marine Bacteria from a Historically Defined Plastic Biodegradation Consortium with the Capacity to Biodegrade Polyhydroxyalkanoates

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Microbe Culture Origins and Revival
2.2. DNA Extraction and Sanger Sequencing of 16S rRNA Genes
2.3. Phylogenetic Analysis
2.4. Isolate Growth on PHAs and Screening for Extracellular PHA Depolymerase Activity
2.5. Whole Genome Sequencing, Assembly, and Annotation
2.6. Pangenomic Analysis
2.7. Analysis of Metabolic Potential
3. Results
3.1. Isolate Identification
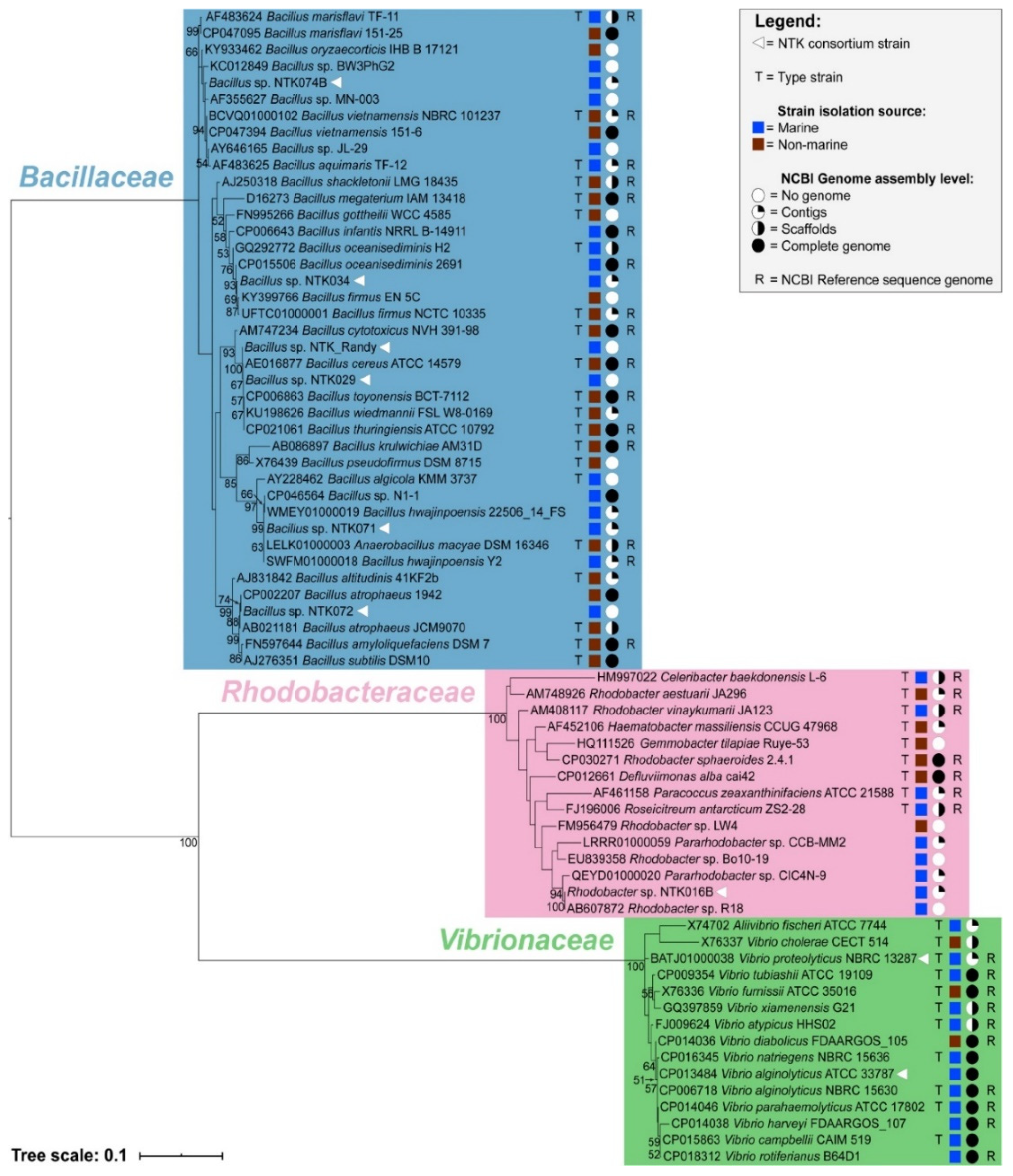
3.2. Phylogenetic Placement of NTK Sequences
3.3. Isolate Growth on PHAs and Screening for Extracellular Depolymerase Activity
3.4. Whole-Genome Sequencing
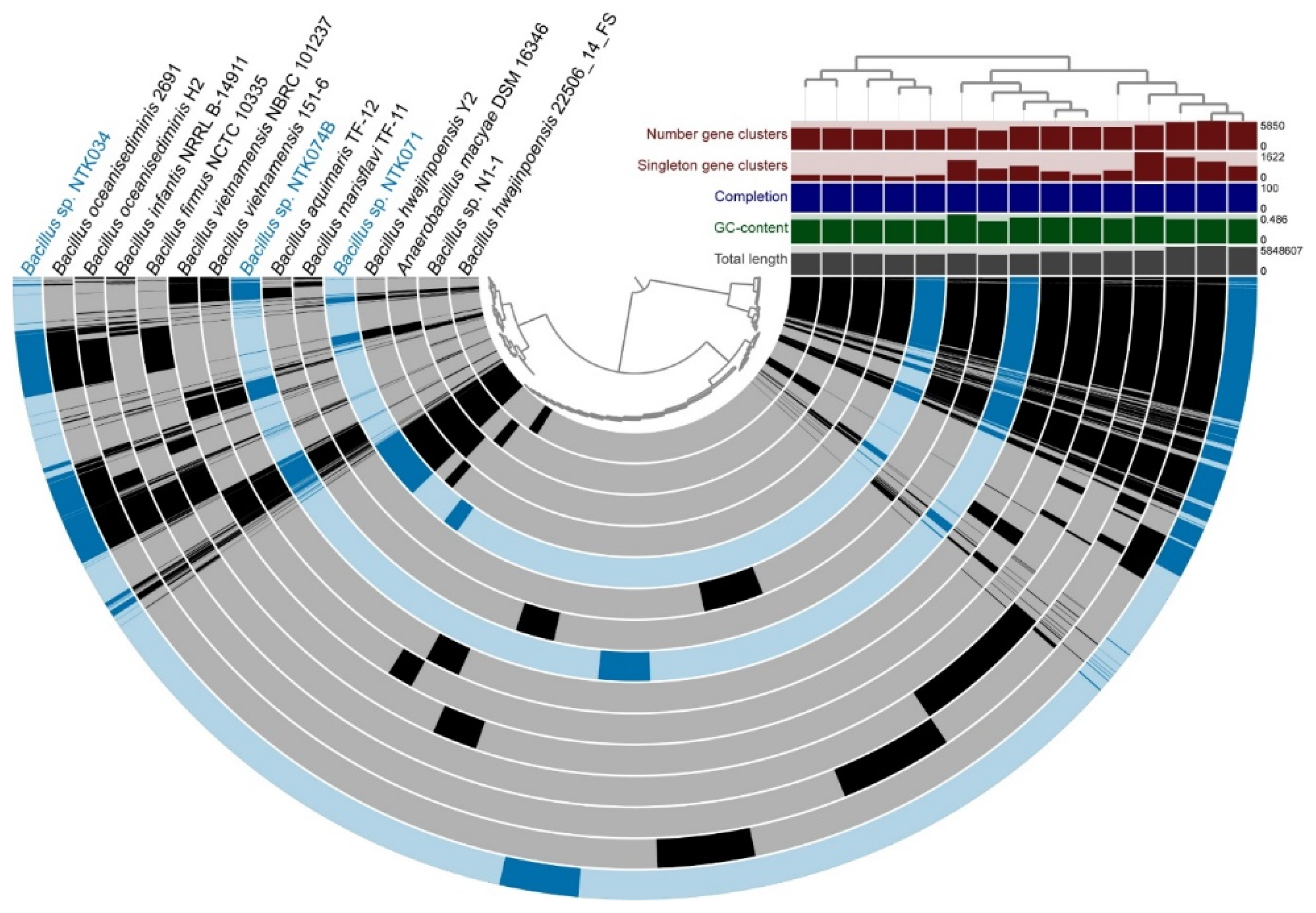
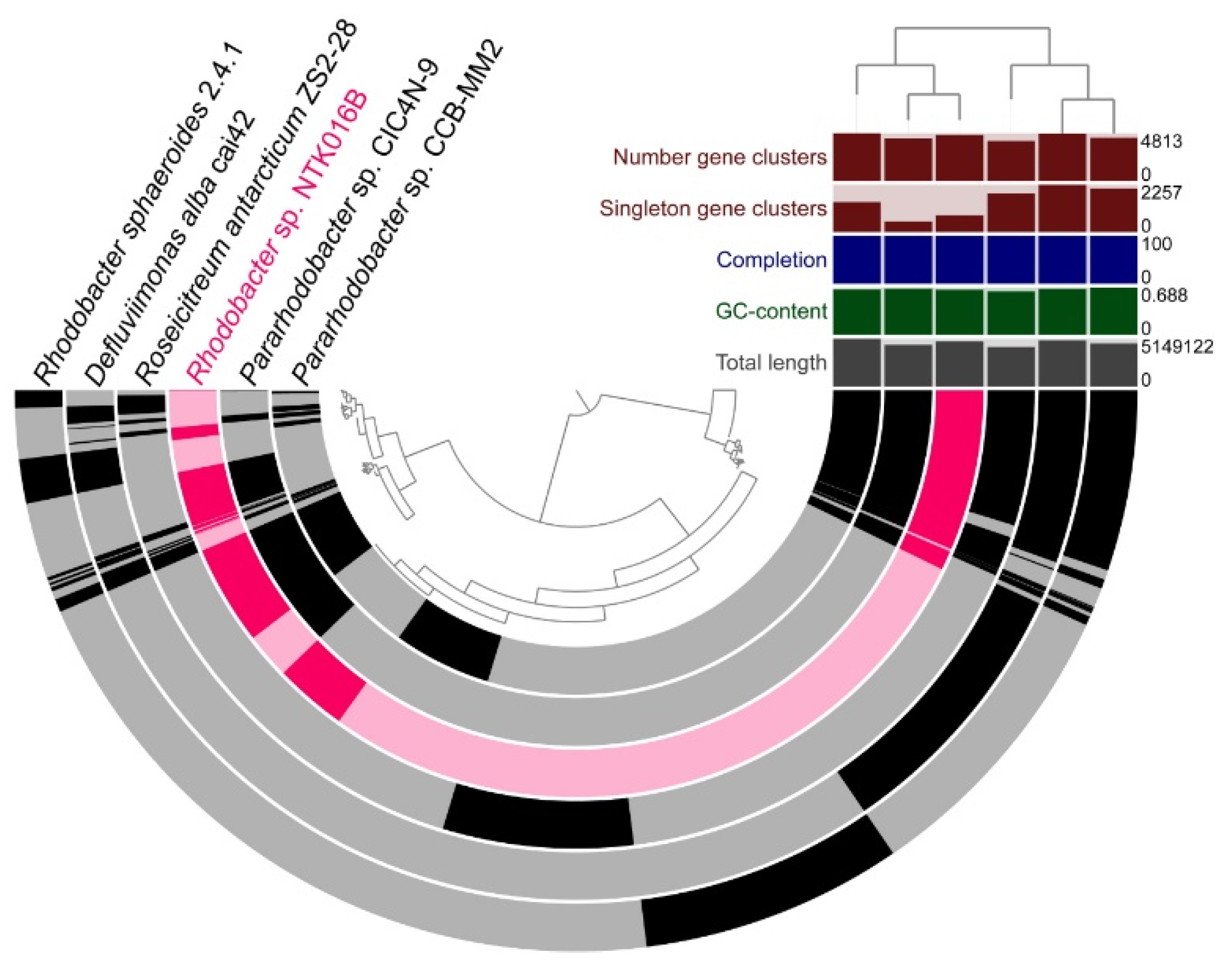
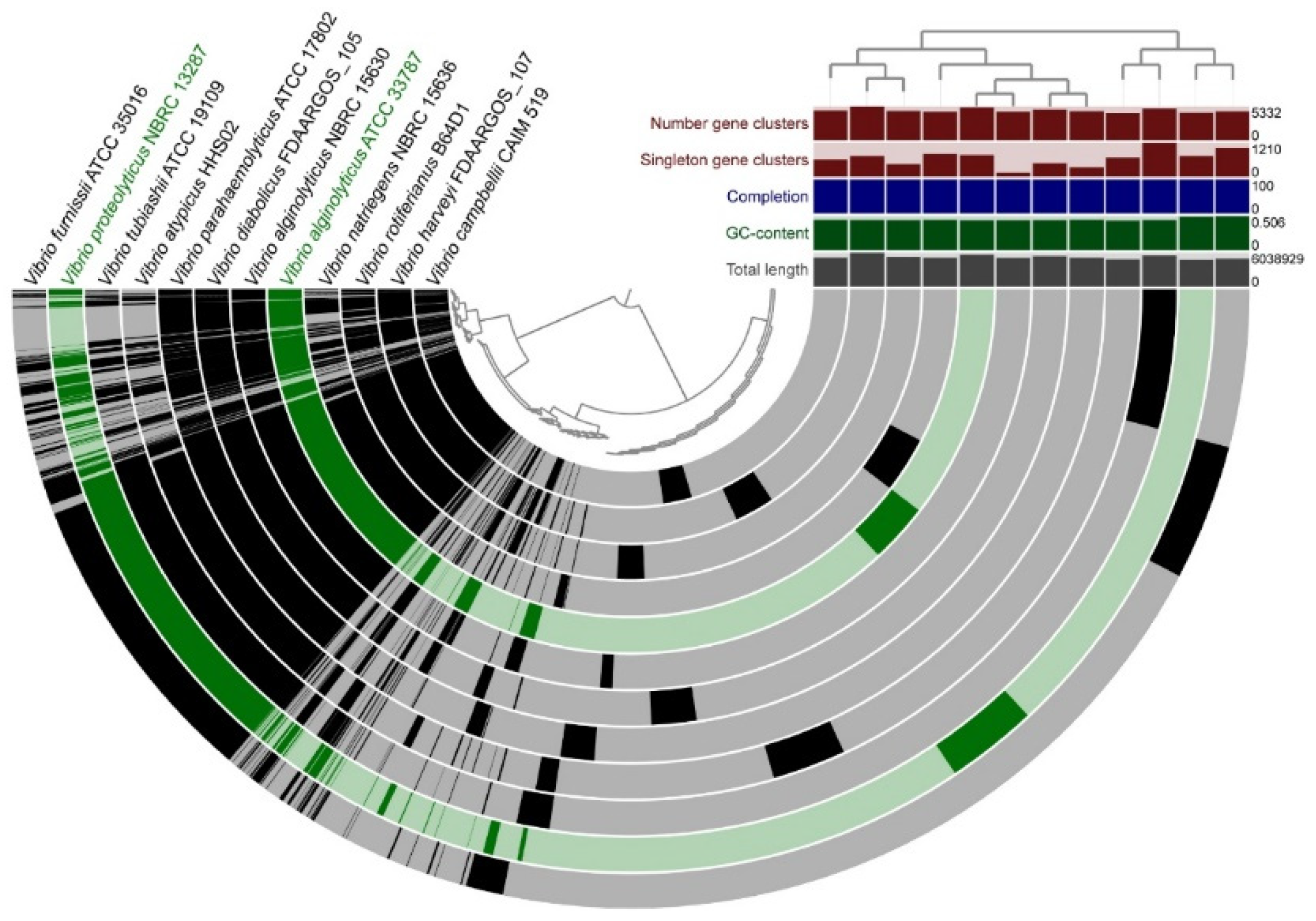
3.5. Pangenomic Analysis
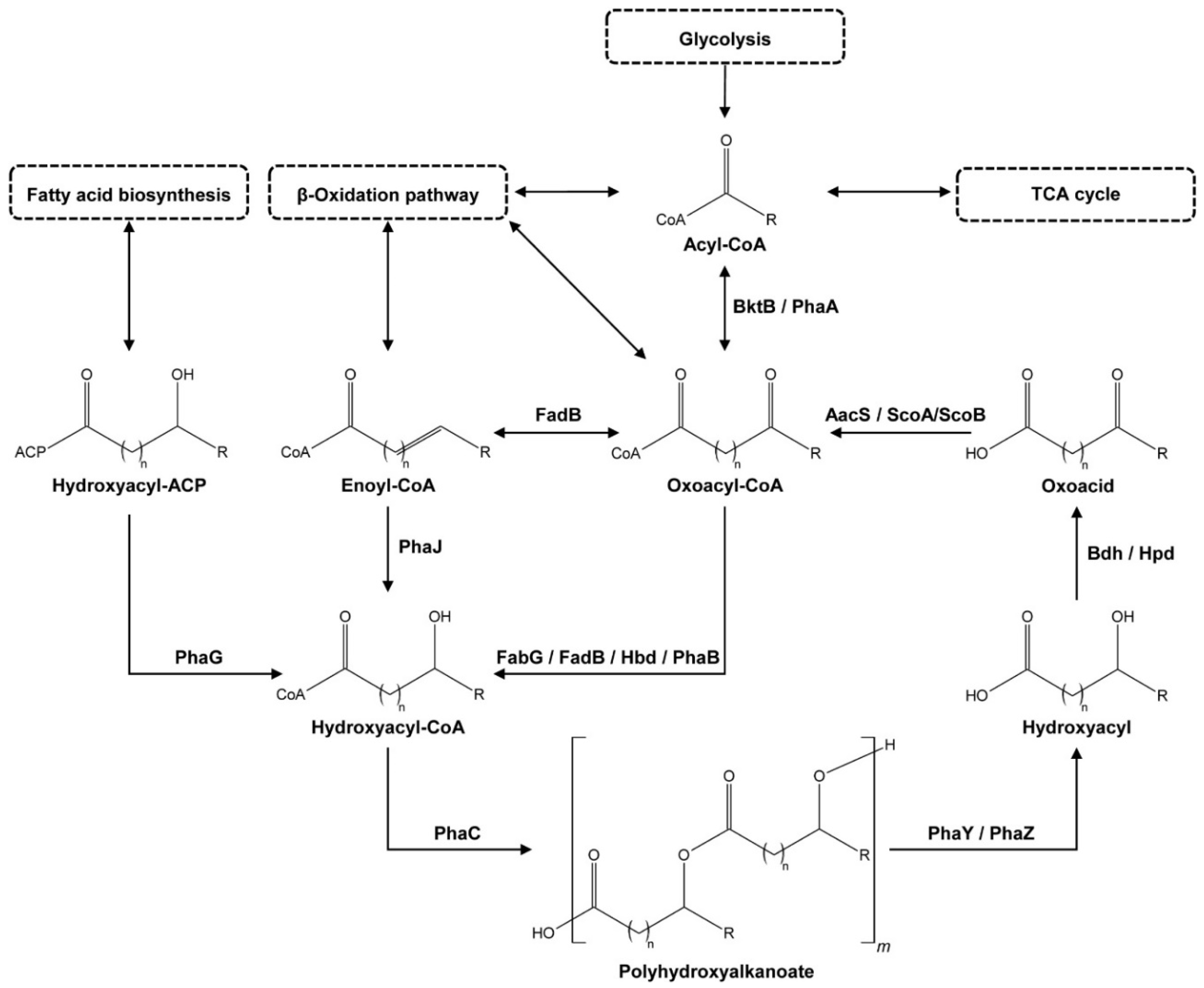
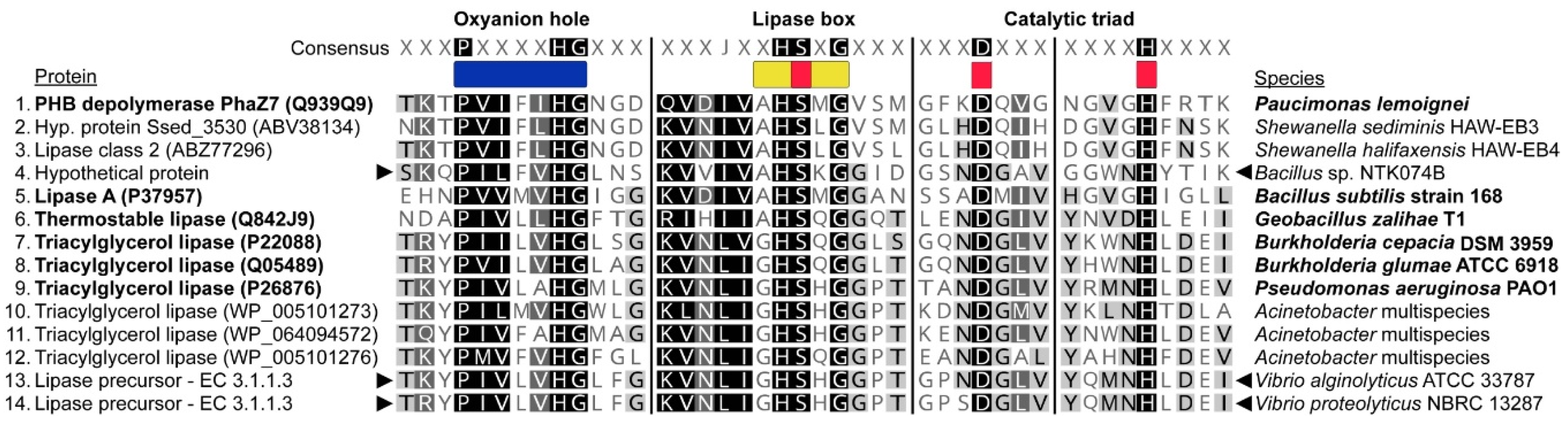
3.6. Analysis of Metabolic Potential
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jambeck, J.R.; Geyer, R.; Wilcox, C.; Siegler, T.R.; Perryman, M.; Andrady, A.; Narayan, R.; Law, K.L. Plastic waste inputs from land into the ocean. Science 2015, 347, 768–771. [Google Scholar] [CrossRef]
- Beaumont, N.J.; Aanesen, M.; Austen, M.C.; Börger, T.; Clark, J.R.; Cole, M.; Hooper, T.; Lindeque, P.K.; Pascoe, C.; Wyles, K.J. Global ecological, social and economic impacts of marine plastic. Mar. Pollut. Bull. 2019, 142, 189–195. [Google Scholar] [CrossRef]
- Henderson, J.R. A pre-and post-MARPOL Annex V summary of Hawaiian monk seal entanglements and marine debris accumulation in the Northwestern Hawaiian Islands, 1982–1998. Mar. Pollut. Bull. 2001, 42, 584–589. [Google Scholar] [CrossRef]
- United Nations. Transforming Our World: The 2030 Agenda for Sustainable Development. Available online: https://sdgs.un.org/2030agenda (accessed on 29 November 2020).
- European Commission. Plastic Waste: A European Strategy to Protect the Planet, Defend Our Citizens and Empower Our Industries; European Commission: Brussels, Belgium, 2018. [Google Scholar]
- European Commission. Circular Economy: New Rules Will Make EU the Global Front-Runner in Waste Management and Recycling; European Commission: Brussels, Belgium, 2018. [Google Scholar]
- European Commission. Circular Economy: Commission Welcomes Council Final Adoption of New Rules on Single-Use Plastics to Reduce Marine Plastic Litter; European Commission: Brussels, Belgium, 2019. [Google Scholar]
- Huang, J.C.; Shetty, A.S.; Wang, M.S. Biodegradable plastics: A review. Adv. Polym. Tech. 1990, 10, 23–30. [Google Scholar] [CrossRef]
- Cózar, A.; Echevarría, F.; González-Gordillo, J.I.; Irigoien, X.; Úbeda, B.; Hernández-León, S.; Palma, Á.T.; Navarro, S.; García-de-Lomas, J.; Ruiz, A. Plastic debris in the open ocean. Proc. Natl. Acad. Sci. USA 2014, 111, 10239–10244. [Google Scholar] [CrossRef]
- Krueger, M.C.; Harms, H.; Schlosser, D. Prospects for microbiological solutions to environmental pollution with plastics. Appl. Microbiol. Biotechnol. 2015, 99, 8857–8874. [Google Scholar] [CrossRef] [PubMed]
- Zumstein, M.T.; Narayan, R.; Kohler, H.-P.E.; McNeill, K.; Sander, M. Dos and do nots when assessing the biodegradation of plastics. Environ. Sci. Technol. 2019, 53, 9967–9969. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Wagner, M. Environmental performance of bio-based and biodegradable plastics: The road ahead. Chem. Soc. Rev. 2017, 46, 6855–6871. [Google Scholar] [CrossRef] [PubMed]
- van Sebille, E.; Aliani, S.; Law, K.L.; Maximenko, N.; Alsina, J.M.; Bagaev, A.; Bergmann, M.; Chapron, B.; Chubarenko, I.; Cózar, A. The physical oceanography of the transport of floating marine debris. Environ. Res. Lett. 2020, 15, 023003. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Hakkarainen, M. Designed to degrade. Science 2017, 358, 872–873. [Google Scholar] [CrossRef]
- Ratto, J.A.; Russo, J.; Allen, A.; Herbert, J.; Wirsen, C. Biodegradable polymers in the marine environment: A tiered approach to assessing microbial degradability. In Biopolymers from Polysaccharides and Agroproteins; Gross, R.A., Scholz, C., Eds.; ACS Publications: Washington, DC, USA, 2001; pp. 316–336. [Google Scholar] [CrossRef]
- Harrison, J.P.; Boardman, C.; O’Callaghan, K.; Delort, A.-M.; Song, J. Biodegradability standards for carrier bags and plastic films in aquatic environments: A critical review. R. Soc. Open Sci. 2018, 5, 171792. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Zettler, L.A.; Zettler, E.R.; Mincer, T.J. Ecology of the plastisphere. Nat. Rev. Microbiol. 2020, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef]
- Castro-Aguirre, E.; Auras, R.; Selke, S.; Rubino, M.; Marsh, T. Insights on the aerobic biodegradation of polymers by analysis of evolved carbon dioxide in simulated composting conditions. Polym. Degrad. Stab. 2017, 137, 251–271. [Google Scholar] [CrossRef]
- Emadian, S.M.; Onay, T.T.; Demirel, B. Biodegradation of bioplastics in natural environments. Waste Manag. 2017, 59, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Dilkes-Hoffman, L.S.; Lant, P.A.; Laycock, B.; Pratt, S. The rate of biodegradation of PHA bioplastics in the marine environment: A meta-study. Mar. Pollut. Bull. 2019, 142, 15–24. [Google Scholar] [CrossRef]
- Greene, J. Marine Biodegradation of PLA, PHA, and Bio-Additive Polyethylene Based on ASTM D7081. California: CalRecycle: 2012. Available online: https://www2.calrecycle.ca.gov/Publications/Details/1435 (accessed on 5 June 2020).
- Sashiwa, H.; Fukuda, R.; Okura, T.; Sato, S.; Nakayama, A. Microbial degradation behavior in seawater of polyester blends containing poly (3-hydroxybutyrate-co-3-hydroxyhexanoate)(PHBHHx). Mar. Drugs 2018, 16, 34. [Google Scholar] [CrossRef]
- Sudesh, K.; Abe, H.; Doi, Y. Synthesis, structure and properties of polyhydroxyalkanoates: Biological polyesters. Prog. Polym. Sci. 2000, 25, 1503–1555. [Google Scholar] [CrossRef]
- Kumar, M.; Rathour, R.; Singh, R.; Sun, Y.; Pandey, A.; Gnansounou, E.; Lin, K.-Y.A.; Tsang, D.C.; Thakur, I.S. Bacterial polyhydroxyalkanoates: Opportunities, challenges, and prospects. J. Clean. Prod. 2020, 121500. [Google Scholar] [CrossRef]
- Choi, S.Y.; Cho, I.J.; Lee, Y.; Kim, Y.J.; Kim, K.J.; Lee, S.Y. Microbial polyhydroxyalkanoates and nonnatural Polyesters. Adv. Mater. 2020, 1907138. [Google Scholar] [CrossRef]
- Zhang, J.; Shishatskaya, E.I.; Volova, T.G.; da Silva, L.F.; Chen, G.-Q. Polyhydroxyalkanoates (PHA) for therapeutic applications. Mater. Sci. Eng. C 2018, 86, 144–150. [Google Scholar] [CrossRef]
- Mudenur, C.; Mondal, K.; Singh, U.; Katiyar, V. Production of polyhydroxyalkanoates and its potential applications. In Advances in Sustainable Polymers; Katiyar, V., Gupta, R.T.G., Eds.; Springer: Singapore, 2019; pp. 131–164. [Google Scholar] [CrossRef]
- Gahlawat, G. Polyhydroxyalkanoates: The future bioplastics. In Polyhydroxyalkanoates Biopolymers; Springer: Cham, Switzerland, 2019; pp. 15–23. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, P.; Ray, S.; Kalia, V.C. Challenges and opportunities for customizing polyhydroxyalkanoates. Indian J. Microbiol. 2015, 55, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Gahlawat, G. Challenges in PHAs production at mass scale. In Polyhydroxyalkanoates Biopolymers; Springer: Cham, Switzerland, 2019; pp. 25–30. [Google Scholar] [CrossRef]
- Tan, G.-Y.A.; Chen, C.-L.; Li, L.; Ge, L.; Wang, L.; Razaad, I.M.N.; Li, Y.; Zhao, L.; Mo, Y.; Wang, J.-Y. Start a research on biopolymer polyhydroxyalkanoate (PHA): A review. Polymers 2014, 6, 706–754. [Google Scholar] [CrossRef]
- Lemoigne, M. Produits de déshydration et de polymérisation de l’acide β-oxybutyrique. Bull. Soc. Chim. Biol. 1926, 8, 770–782. [Google Scholar]
- Chen, G.-Q.; Chen, X.-Y.; Wu, F.-Q.; Chen, J.-C. Polyhydroxyalkanoates (PHA) toward cost competitiveness and functionality. Adv. Ind. Eng. Polym. Res. 2020, 3, 1–7. [Google Scholar] [CrossRef]
- Bresan, S.; Sznajder, A.; Hauf, W.; Forchhammer, K.; Pfeiffer, D.; Jendrossek, D. Polyhydroxyalkanoate (PHA) granules have no phospholipids. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Jendrossek, D.; Handrick, R. Microbial degradation of polyhydroxyalkanoates. Annu. Rev. Microbiol. 2002, 56, 403–432. [Google Scholar] [CrossRef]
- Kobayashi, T.; Shiraki, M.; Abe, T.; Sugiyama, A.; Saito, T. Purification and properties of an intracellular 3-hydroxybutyrate-oligomer hydrolase (PhaZ2) in Ralstonia eutropha H16 and its identification as a novel intracellular poly (3-hydroxybutyrate) depolymerase. J. Bacteriol. 2003, 185, 3485–3490. [Google Scholar] [CrossRef]
- Jendrossek, D. Extracellular Polyhydroxyalkanoate (PHA) Depolymerases: The Key Enzymes of PHA Degradation. In Biopolymers; Steinbüchel, A., Ed.; Wiley-VCH: Weinheim, Germany, 2005; Volume 3. [Google Scholar]
- Sznajder, A.; Pfeiffer, D.; Jendrossek, D. Comparative proteome analysis reveals four novel polyhydroxybutyrate (PHB) granule-associated proteins in Ralstonia eutropha H16. Appl. Environ. Microbiol. 2015, 81, 1847–1858. [Google Scholar] [CrossRef][Green Version]
- Carr, P.D.; Ollis, D.L. α/βHydrolase fold: An update. Protein Pept. Lett. 2009, 16, 1137–1148. [Google Scholar] [CrossRef]
- Oh, C.; Kim, T.D.; Kim, K.K. Carboxylic ester hydrolases in bacteria: Active site, structure, function and application. Crystals 2019, 9, 597. [Google Scholar] [CrossRef]
- Knoll, M.; Hamm, T.M.; Wagner, F.; Martinez, V.; Pleiss, J. The PHA Depolymerase Engineering Database: A systematic analysis tool for the diverse family of polyhydroxyalkanoate (PHA) depolymerases. BMC Bioinform. 2009, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shiraki, M.; Saito, T. Purification of an extracellular D-(-)-3-hydroxybutyrate oligomer hydrolase from Pseudomonas sp. strain A1 and cloning and sequencing of its gene. J. Bacteriol. 1997, 179, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Kobayashi, T.; Shiraki, M.; Saito, T. Roles of poly (3-hydroxybutyrate) depolymerase and 3HB-oligomer hydrolase in bacterial PHB metabolism. Curr. Microbiol. 2004, 48, 424–427. [Google Scholar] [CrossRef]
- Kobayashi, T.; Uchino, K.; Abe, T.; Yamazaki, Y.; Saito, T. Novel intracellular 3-hydroxybutyrate-oligomer hydrolase in Wautersia eutropha H16. J. Bacteriol. 2005, 187, 5129–5135. [Google Scholar] [CrossRef]
- Lu, J.; Takahashi, A.; Ueda, S. 3-Hydroxybutyrate oligomer hydrolase and 3-hydroxybutyrate dehydrogenase participate in intracellular polyhydroxybutyrate and polyhydroxyvalerate degradation in Paracoccus denitrificans. Appl. Environ. Microbiol. 2014, 80, 986–993. [Google Scholar] [CrossRef]
- Delafield, F.; Doudoroff, M.; Palleroni, N.; Lusty, C.; Contopoulos, R. Decomposition of poly-β-hydroxybutyrate by pseudomonads. J. Bacteriol. 1965, 90, 1455–1466. [Google Scholar] [CrossRef]
- Aneja, P.; Charles, T. Poly-3-hydroxybutyrate degradation in Rhizobium (Sinorhizobium) meliloti: Isolation and characterization of a gene encoding 3-hydroxybutyrate dehydrogenase. J. Bacteriol. 1999, 181, 849–857. [Google Scholar] [CrossRef]
- Den, H.; Robinson, W.G.; Coon, M.J. Enzymatic conversion of β-hydroxypropionate to malonic semialdehyde. J. Biol. Chem. 1959, 234, 1666–1671. [Google Scholar] [CrossRef]
- Otzen, C.; Bardl, B.; Jacobsen, I.D.; Nett, M.; Brock, M. Candida albicans utilizes a modified β-oxidation pathway for the degradation of toxic propionyl-CoA. J. Biol. Chem. 2014, 289, 8151–8169. [Google Scholar] [CrossRef]
- Stols, L.; Zhou, M.; Eschenfeldt, W.H.; Millard, C.S.; Abdullah, J.; Collart, F.R.; Kim, Y.; Donnelly, M.I. New vectors for co-expression of proteins: Structure of Bacillus subtilis ScoAB obtained by high-throughput protocols. Protein Expr. Purif. 2007, 53, 396–403. [Google Scholar] [CrossRef]
- Tucker, A.C.; Escalante-Semerena, J.C. Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol. Biotechnol. 2013, 87, 152–167. [Google Scholar] [CrossRef]
- ASTM International. ASTM D6691-01, Standard Test Method for Determining Aerobic Biodegradation of Plastic Materials in the Marine Environment by a Defined Microbial Consortium; ASTM International: West Conshohocken, PA, USA, 2001; p. 4. [Google Scholar] [CrossRef]
- McCassie, J.; Mayer, J.; Stote, R.; Shupe, A.; Stenhouse, P.; Dell, P.; Kaplan, D. Biodegradation kinetics in marine and soil systems. In Biodegradable Polymers and Packaging; Ching, C., Kaplan, D., Thomas, E., Eds.; Technomic Publishing Company Inc.: Landcaster, PA, USA, 1993; pp. 247–256. [Google Scholar]
- Mayer, J.; Kaplan, D.; Stote, R.; Dixon, K.; Shupe, A.; Allen, A.; McCassie, J. Biodegradation of polymer films in marine and soil environments. In Hydrogels and Biodegradable Polymers for Bioapplications; Ottenbrite, R., Huang, S., Park, K., Eds.; ACS Publications: Washington, DC, USA, 1996; pp. 159–170. [Google Scholar] [CrossRef]
- Allen, A.L.; Mayer, J.; Stote, R.; Kaplan, D.L. Simulated marine respirometry of biodegradable polymers. J. Environ. Polym. Degr. 1994, 2, 237–244. [Google Scholar] [CrossRef]
- Spence, K.E.; Allen, A.L.; Wang, S.; Jane, J. Soil and Marine Biodegradation of Protein—Starch Plastics. In Hydrogels and Biodegradable Polymers for Bioapplications; Ottenbrite, R., Huang, S., Park, K., Eds.; ACS Publications: Washington, DC, USA, 1996; pp. 149–158. [Google Scholar] [CrossRef]
- Thellen, C.; Coyne, M.; Froio, D.; Auerbach, M.; Wirsen, C.; Ratto, J.A. A processing, characterization and marine biodegradation study of melt-extruded polyhydroxyalkanoate (PHA) films. J. Polym. Environ. 2008, 16, 1–11. [Google Scholar] [CrossRef]
- Merkel, J.R.; Traganza, E.D.; Mukherjee, B.B.; Griffin, T.B.; Prescott, J. Proteolytic activity and general characteristics of a marine bacterium, Aeromonas proteolytica sp. n. J. Bacteriol. 1964, 87, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Baumann, L.; Mandel, M. Taxonomy of marine bacteria: The genus Beneckea. J. Bacteriol. 1971, 107, 268–294. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Guillard, R.R.; Ryther, J.H. Studies of marine planktonic diatoms: I. Cyclotella nana Hustedt, and Detonula confervacea (Cleve) Gran. Can. J. Microbiol. 1962, 8, 229–239. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genome 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ. 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Delmont, T.O.; Eren, A.M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ. 2018, 6, e4320. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H. Predicting secretory proteins with SignalP. In Protein Function Prediction; Kihara, D., Ed.; Humana Press: New York, NY, USA, 2017; pp. 59–73. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Lees, J.; Yeats, C.; Perkins, J.; Sillitoe, I.; Rentzsch, R.; Dessailly, B.H.; Orengo, C. Gene3D: A domain-based resource for comparative genomics, functional annotation and protein network analysis. Nucleic Acids Res. 2012, 40, D465–D471. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Attwood, T.K.; Coletta, A.; Muirhead, G.; Pavlopoulou, A.; Philippou, P.B.; Popov, I.; Roma-Mateo, C.; Theodosiou, A.; Mitchell, A.L. The PRINTS database: A fine-grained protein sequence annotation and analysis resource—its status in 2012. Database 2012, 2012. [Google Scholar] [CrossRef]
- Oates, M.E.; Stahlhacke, J.; Vavoulis, D.V.; Smithers, B.; Rackham, O.J.; Sardar, A.J.; Zaucha, J.; Thurlby, N.; Fang, H.; Gough, J. The SUPERFAMILY 1.75 database in 2014: A doubling of data. Nucleic Acids Res. 2015, 43, D227–D233. [Google Scholar] [CrossRef]
- Handrick, R.; Reinhardt, S.; Focarete, M.L.; Scandola, M.; Adamus, G.; Kowalczuk, M.; Jendrossek, D. A new type of thermoalkalophilic hydrolase of Paucimonas lemoignei with high specificity for amorphous polyesters of short chain-length hydroxyalkanoic acids. J. Biol. Chem. 2001, 276, 36215–36224. [Google Scholar] [CrossRef]
- Sharma, P.K.; Mohanan, N.; Sidhu, R.; Levin, D.B. Colonization and degradation of polyhydroxyalkanoates by lipase-producing bacteria. Can. J. Microbiol. 2019, 65, 461–475. [Google Scholar] [CrossRef]
- Sharifah, E.N.; Eguchi, M. The phytoplankton Nannochloropsis oculata enhances the ability of Roseobacter clade bacteria to inhibit the growth of fish pathogen Vibrio anguillarum. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Jeong, H.; Kim, H.J.; Lee, D.-W.; Lee, S.J. Complete genome sequence of Bacillus oceanisediminis 2691, a reservoir of heavy-metal resistance genes. Mar. Genome 2016, 30, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrn DB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhao, Y.; Wang, X.; Qu, C.; Miao, J. Complete genome sequence of Bacillus sp. N1-1, a κ-selenocarrageenan degrading bacterium isolated from the cold seep in the South China Sea. Mar. Genome 2020, 100771. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ding, Z.; Ji, Y.; Zhao, J.; Liu, X.; Tian, J.; Wu, N.; Fan, Y. An operon consisting of a P-type ATPase gene and a transcriptional regulator gene responsible for cadmium resistances in Bacillus vietamensis 151–6 and Bacillus marisflavi 151–25. BMC Microbiol. 2020, 20, 18. [Google Scholar] [CrossRef]
- Lai, Q.; Liu, X.; Yuan, J.; Xie, S.; Shao, Z. Pararhodobacter marinus sp. nov., isolated from deep-sea water of the Indian Ocean. Int. J. Syst. Evol. Microbiol. 2019, 69, 932–936. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Hermawan, S.; Singh, C.B.; Jendrossek, D. Structural basis of poly (3-hydroxybutyrate) hydrolysis by PhaZ7 depolymerase from Paucimonas lemoignei. J. Mol. Biol. 2008, 382, 1184–1194. [Google Scholar] [CrossRef]
- Wakadkar, S.; Hermawan, S.; Jendrossek, D.; Papageorgiou, A.C. The structure of PhaZ7 at atomic (1.2 Å) resolution reveals details of the active site and suggests a substrate-binding mode. Acta Cryst. 2010, F66, 648–654. [Google Scholar] [CrossRef]
- Jendrossek, D.; Hermawan, S.; Subedi, B.; Papageorgiou, A.C. Biochemical analysis and structure determination of Paucimonas lemoignei poly (3-hydroxybutyrate) (PHB) depolymerase PhaZ 7 muteins reveal the PHB binding site and details of substrate–enzyme interactions. Mol. Biotechnol. 2013, 90, 649–664. [Google Scholar] [CrossRef]
- Rüger, H.-J.; Krambeck, H.-J. Evaluation of the BIOLOG substrate metabolism system for classification of marine bacteria. Syst. Appl. Microbiol. 1994, 17, 281–288. [Google Scholar] [CrossRef]
- Liu, Y.; Du, J.; Lai, Q.; Zeng, R.; Ye, D.; Xu, J.; Shao, Z. Proposal of nine novel species of the Bacillus cereus group. Int. J. Syst. Evol. Microbiol. 2017, 67, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Jacquin, J.; Cheng, J.; Odobel, C.; Pandin, C.; Conan, P.; Pujo-Pay, M.; Barbe, V.; Meistertzheim, A.-L.; Ghiglione, J.-F. Microbial ecotoxicology of marine plastic debris: A review on colonization and biodegradation by the “plastisphere”. Front. Microbiol. 2019, 10, 865. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Lovell, C.R. Numerical dominance and phylotype diversity of marine Rhodobacter species during early colonization of submerged surfaces in coastal marine waters as determined by 16S ribosomal DNA sequence analysis and fluorescence in situ hybridization. Appl. Environ. Microbiol. 2002, 68, 496–504. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dang, H.; Li, T.; Chen, M.; Huang, G. Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl. Environ. Microbiol. 2008, 74, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Elifantz, H.; Horn, G.; Ayon, M.; Cohen, Y.; Minz, D. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean coastal seawater. FEMS Microbiol. Ecol. 2013, 85, 348–357. [Google Scholar] [CrossRef]
- Zettler, E.R.; Mincer, T.J.; Amaral-Zettler, L.A. Life in the “plastisphere”: Microbial communities on plastic marine debris. Environ. Sci. Technol. 2013, 47, 7137–7146. [Google Scholar] [CrossRef]
- De Tender, C.A.; Devriese, L.I.; Haegeman, A.; Maes, S.; Ruttink, T.; Dawyndt, P. Bacterial community profiling of plastic litter in the Belgian part of the North Sea. Environ. Sci. Technol. 2015, 49, 9629–9638. [Google Scholar] [CrossRef]
- Pinnell, L.J.; Turner, J.W. Shotgun metagenomics reveals the benthic microbial community response to plastic and bioplastic in a coastal marine environment. Front. Microbiol. 2019, 10, 1252. [Google Scholar] [CrossRef]
- Kirstein, I.V.; Kirmizi, S.; Wichels, A.; Garin-Fernandez, A.; Erler, R.; Löder, M.; Gerdts, G. Dangerous hitchhikers? Evidence for potentially pathogenic Vibrio spp. on microplastic particles. Mar. Environ. Res. 2016, 120, 1–8. [Google Scholar] [CrossRef]
- Dussud, C.; Meistertzheim, A.; Conan, P.; Pujo-Pay, M.; George, M.; Fabre, P.; Coudane, J.; Higgs, P.; Elineau, A.; Pedrotti, M. Evidence of niche partitioning among bacteria living on plastics, organic particles and surrounding seawaters. Environ. Pollut. 2018, 236, 807–816. [Google Scholar] [CrossRef]
- Viljakainen, V.R.; Hug, L.A. The phylogenetic and global distribution of extracellular bioplastic degrading genes. BioRxiv 2020. [Google Scholar] [CrossRef]
- Li, Z.; Lin, H.; Ishii, N.; Chen, G.-Q.; Inoue, Y. Study of enzymatic degradation of microbial copolyesters consisting of 3-hydroxybutyrate and medium-chain-length 3-hydroxyalkanoates. Polym. Degrad. Stab. 2007, 92, 1708–1714. [Google Scholar] [CrossRef]
- Weng, Y.-X.; Wang, X.-L.; Wang, Y.-Z. Biodegradation behavior of PHAs with different chemical structures under controlled composting conditions. Polym. Test 2011, 30, 372–380. [Google Scholar] [CrossRef]
- Altaee, N.; El-Hiti, G.A.; Fahdil, A.; Sudesh, K.; Yousif, E. Biodegradation of different formulations of polyhydroxybutyrate films in soil. SpringerPlus 2016, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Volova, T.G.; Prudnikova, S.V.; Vinogradova, O.N.; Syrvacheva, D.A.; Shishatskaya, E.I. Microbial degradation of polyhydroxyalkanoates with different chemical compositions and their biodegradability. Microb. Ecol. 2017, 73, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Yoshie, N.; Nakasato, K.; Fujiwara, M.; Kasuya, K.; Abe, H.; Doi, Y.; Inoue, Y. Effect of low molecular weight additives on enzymatic degradation of poly (3-hydroxybutyrate). Polymer 2000, 41, 3227–3234. [Google Scholar] [CrossRef]
- Abe, H.; Doi, Y. Side-chain effect of second monomer units on crystalline morphology, thermal properties, and enzymatic degradability for random copolyesters of (R)-3-hydroxybutyric acid with (R)-3-hydroxyalkanoic acids. Biomacromolecules 2002, 3, 133–138. [Google Scholar] [CrossRef]
- Ishida, K.; Asakawa, N.; Inoue, Y. Structure, Properties and Biodegradation of Some Bacterial Copoly (Hydroxyalkanoate)s. In Proceedings of the Macromolecular Symposia, Seoul, Korea, 1–4 June 2004; pp. 47–58. [Google Scholar] [CrossRef]
- Numata, K.; Kikkawa, Y.; Tsuge, T.; Iwata, T.; Doi, Y.; Abe, H. Enzymatic degradation processes of poly [(R)-3-hydroxybutyric acid] and poly [(R)-3-hydroxybutyric acid-co-(R)-3-hydroxyvaleric acid] single crystals revealed by atomic force microscopy: Effects of molecular weight and second-monomer composition on erosion rates. Biomacromolecules 2005, 6, 2008–2016. [Google Scholar] [CrossRef]
- Zhang, J.; Kasuya, K.; Hikima, T.; Takata, M.; Takemura, A.; Iwata, T. Mechanical properties, structure analysis and enzymatic degradation of uniaxially cold-drawn films of poly [(R)-3-hydroxybutyrate-co-4-hydroxybutyrate]. Polym. Degrad. Stab. 2011, 96, 2130–2138. [Google Scholar] [CrossRef]
- Anbukarasu, P.; Sauvageau, D.; Elias, A. Tuning the properties of polyhydroxybutyrate films using acetic acid via solvent casting. Sci. Rep. 2015, 5, 17884. [Google Scholar] [CrossRef]
- Ma, W.-T.; Lin, J.-H.; Chen, H.-J.; Chen, S.-Y.; Shaw, G.-C. Identification and characterization of a novel class of extracellular poly (3-hydroxybutyrate) depolymerase from Bacillus sp. strain NRRL B-14911. Appl. Environ. Microbiol. 2011, 77, 7924–7932. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, K.-E.; Steinbüchel, A.; Jendrossek, D. Substrate specificities of bacterial polyhydroxyalkanoate depolymerases and lipases: Bacterial lipases hydrolyze poly (omega-hydroxyalkanoates). Appl. Environ. Microbiol. 1995, 61, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Doi, Y.; Sema, Y.; Tomita, K. Substrate specificities in hydrolysis of polyhydroxyalkanoates by microbial esterases. Biotechnol. Lett. 1993, 15, 601–604. [Google Scholar] [CrossRef]
- Mohamed, R.A.; Salleh, A.B.; Leow, A.T.C.; Yahaya, N.M.; Rahman, M.B.A. Ability of T1 lipase to degrade amorphous P(3HB): Structural and functional study. Mol. Biotechnol. 2017, 59, 284–293. [Google Scholar] [CrossRef]
- Yao, T.; Xu, L.; Ying, H.; Huang, H.; Yan, M. The catalytic property of 3-hydroxyisobutyrate dehydrogenase from Bacillus cereus on 3-hydroxypropionate. Appl. Biochem. Biotechnol. 2010, 160, 694–703. [Google Scholar] [CrossRef]
- Park, S.C.; Kim, P.-H.; Lee, G.-S.; Kang, S.G.; Ko, H.-J.; Yoon, S.-I. Structural and biochemical characterization of the Bacillus cereus 3-hydroxyisobutyrate dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 474, 522–527. [Google Scholar] [CrossRef]
- Zhou, S.; Raj, S.M.; Ashok, S.; Edwardraja, S.; Lee, S.-g.; Park, S. Cloning, expression and characterization of 3-hydroxyisobutyrate dehydrogenase from Pseudomonas denitrificans ATCC 13867. PLoS ONE 2013, 8, e62666. [Google Scholar] [CrossRef][Green Version]
- Lee, P.; Raj, S.M.; Zhou, S.; Ashok, S.; Edwardraja, S.; Park, S. 3-Hydroxyisobutyrate dehydrogenase-I from Pseudomonas denitrificans ATCC 13867 degrades 3-hydroxypropionic acid. Biotechnol. Bioproc. E 2014, 19, 1–7. [Google Scholar] [CrossRef]
- Chowdhury, E.K.; Nagata, S.; Misono, H. 3-Hydroxyisobutyrate dehydrogenase from Pseudomonas putida E23: Purification and characterization. Biosci. Biotechnol. Biochem. 1996, 60, 2043–2047. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Original Identity | Isolate Fate | 16S rRNA Gene BLASTn Hit | Coverage | Identity | Accession |
|---|---|---|---|---|---|---|
| NTK009 | Unknown | Lost | - | - | - | - |
| NTK016B | Unknown | Revived | Rhodobacter sp. R18 | 99.72% | 97% | AB607872 |
| NTK029 | Pseudoalteromonas haloplanktis | Revived | Bacillus cereus group | 100% | 99.93% | - |
| NTK034 | Unknown | Revived | Bacillus oceanisediminis 2691 | 100% | 99.87% | CP015506 |
| NTK039 | Pseudomonas creosotensis | Lost | - | - | - | - |
| NTK045 | Vibrio proteolyticus | Replaced by ATCC 15338 | Vibrio proteolyticus ATCC 15338 = NBRC 13287 | 100% | 99.64% | NR_026128 |
| NTK049 | Vibrio alginolyticus | Replaced by ATCC 33787 | Vibrio alginolyticus ATCC 33787 | 100% | 99.35% | CP013484 |
| NTK060 | Vibrio campbellii | Lost | - | - | - | - |
| NTK071 | Bacillus megaterium | Revived | Bacillus sp. N1-1 | 100% | 99.49% | CP046564 |
| NTK072 | Vibrio furnissii | Revived | Bacillus atrophaeus, multiple strains | 100% | 100% | - |
| NTK073 | Xanthomas campestris | Lost | - | - | - | - |
| NTK074Act | Actinomycete sp. | Lost | - | - | - | - |
| NTK074B | Unknown | Revived | Bacillus vietnamensis 151-6 | 100% | 99.68% | CP047394 |
| NTK_Randy | Bacillus sp. | Revived | Bacillus cereus group | 100% | 99.93% | - |
| Isolate | 16S rRNA Gene Top Blastn Hit | Growth on PHAs | Depolymerase Activity | Sequenced Genome |
|---|---|---|---|---|
| ATCC 15338 | V. proteolyticus ATCC 15338 = NBRC 13287 | N.D. a | Yes | Existing |
| ATCC 33787 | V. alginolyticus ATCC 33787 | N.D. a | No | Existing |
| NTK016B | Rhodobacter sp. R18 | No | No | This study |
| NTK029 | B. cereus group | No | N.D. | No |
| NTK034 | B. oceanisediminis 2691 | No | No | This study |
| NTK071 | Bacillus sp. N1-1 | No | No | This study |
| NTK072 | B. atrophaeus, multiple strains | No | Yes | No |
| NTK074B | B. vietnamensis 151-6 | Yes | Yes | This study |
| NTK_Randy | B. cereus group | Yes | No | No |
| Attribute | NTK016B | NTK034 | NTK071 | NTK074B |
|---|---|---|---|---|
| Genome size (bp) | 4,854,159 | 5,599,963 | 4,164,462 | 4,250,699 |
| GC-content (%) | 65.4 | 41.0 | 39.9 | 43.5 |
| Number of contigs | 162 | 100 | 229 | 446 |
| N50 | 231,698 | 423,522 | 225,829 | 300,122 |
| L50 | 8 | 4 | 5 | 5 |
| Average coverage (reads) | 63.8 | 51.8 | 51.7 | 74.0 |
| Number of coding sequences | 4889 | 6087 | 4385 | 4852 |
| Number of RNAs | 46 | 127 | 102 | 128 |
| Function | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Gene | phaA/bktB | fabG | fadB | hbd | phaB | phaC | (e) phaY | (e) phaZ | (i) phaZ | bdh | aacS | scoA and scoB |
| Bacillus sp. NTK034 ◄ | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus oceanisediminis 2691 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus oceanisediminis H2 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus infantis NRRL B-14911 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | ✓ | ✓ | ✓ | - | ✓ | |
| Bacillus firmus NCTC 10335 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus vietnamensis NBRC 101237 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | ✓ | ✓ | - | ✓ | |
| Bacillus vietnamensis 151-6 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | ✓ | ✓ | - | ✓ | |
| Bacillus sp. NTK074B ◄ | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus aquimaris TF-12 | ✓ | ✓ | ✓ | ✓ | - | - | - | - | ✓ | ✓ | - | ✓ | |
| Bacillus marisflavi TF-11 | ✓ | ✓ | ✓ | ✓ | - | ✓ | - | - | - | ✓ | - | ✓ | |
| Bacillus sp. NTK071 ◄ | ✓ | ✓ | ✓ | ✓ | - | - | - | - | - | ✓ | - | ✓ | |
| Bacillus hwajinpoensis Y2 | ✓ | ✓ | ✓ | ✓ | - | - | - | - | - | ✓ | - | ✓ | |
| Anaerobacillus macyae DSM 16346 | ✓ | ✓ | ✓ | ✓ | - | - | - | ✓ | ✓ | ✓ | - | ✓ | |
| Bacillus sp. N1-1 | ✓ | ✓ | ✓ | ✓ | - | - | - | - | - | ✓ | - | ✓ | |
| Bacillus hwajinpoensis 22506_14_FS | ✓ | ✓ | ✓ | ✓ | - | - | - | - | - | ✓ | - | ✓ | |
| Rhodobacter sphaeroides 2.4.1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | ✓ a | ✓ | ✓ | - | ✓ | |
| Defluviimonas alba cai42 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | - | ✓ | |
| Roseicitreum antarcticum ZS2-28 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | - | ✓ | |
| Rhodobacter sp. NTK016B ◄ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | ✓ | ✓ | |
| Pararhodobacter sp. CIC4N-9 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | ✓ | ✓ | |
| Pararhodobacter sp. CCB-MM2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | ✓ | ✓ | |
| Vibrio furnissii ATCC 35016 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio proteolyticus NBRC 13287 ◄ | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio tubiashii ATCC 19109 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio atypicus HHS02 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio parahaemolyticus ATCC 17802 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio diabolicus FDAARGOS_105 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio alginolyticus NBRC 15630 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio alginolyticus ATCC 33787 ◄ | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio natriegens NBRC 15636 | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ | - | - | - | ✓ | - | |
| Vibrio rotiferianus B64D1 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio harveyi FDAARGOS_107 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Vibrio campbellii CAIM 519 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | - | - | ✓ | - | |
| Function: | |||||||||||||
| 1 | Acetyl-CoA acetyltransferase/β-ketothiolase (EC 2.3.1.9 and EC 2.3.1.16) | 7 | (extracellular) PHA oligomer hydrolase (EC 3.1.1.22) | ||||||||||
| 2 | 3-oxoacyl-[acyl-carrier-protein] reductase (EC 1.1.1.100 with EC 1.1.1.36 capacity) | 8 | (extracellular) PHA depolymerase (EC 3.1.1.75 and EC 3.1.1.76) | ||||||||||
| 3 | Enoyl-CoA hydratase (EC 4.2.1.17)/ Hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35) | 9 | (intracellular) PHA depolymerase (EC 3.1.1.75 and EC 3.1.1.76) | ||||||||||
| 4 | 3-hydroxybutyryl-CoA dehydrogenase (EC 1.1.1.157) | 10 | 3-hydroxybutyrate dehydrogenase (EC 1.1.1.30) | ||||||||||
| 5 | Acetoacetyl-CoA reductase (EC 1.1.1.36) | 11 | Acetoacetyl-CoA synthetase (EC 6.2.1.16) | ||||||||||
| 6 | PHA synthase (EC 2.3.1.-) | 12 | 3-oxoacid CoA-transferase subunit A and B (EC 2.8.3.5) | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Vogel, F.A.; Schlundt, C.; Stote, R.E.; Ratto, J.A.; Amaral-Zettler, L.A. Comparative Genomics of Marine Bacteria from a Historically Defined Plastic Biodegradation Consortium with the Capacity to Biodegrade Polyhydroxyalkanoates. Microorganisms 2021, 9, 186. https://doi.org/10.3390/microorganisms9010186
de Vogel FA, Schlundt C, Stote RE, Ratto JA, Amaral-Zettler LA. Comparative Genomics of Marine Bacteria from a Historically Defined Plastic Biodegradation Consortium with the Capacity to Biodegrade Polyhydroxyalkanoates. Microorganisms. 2021; 9(1):186. https://doi.org/10.3390/microorganisms9010186
Chicago/Turabian Stylede Vogel, Fons A., Cathleen Schlundt, Robert E. Stote, Jo Ann Ratto, and Linda A. Amaral-Zettler. 2021. "Comparative Genomics of Marine Bacteria from a Historically Defined Plastic Biodegradation Consortium with the Capacity to Biodegrade Polyhydroxyalkanoates" Microorganisms 9, no. 1: 186. https://doi.org/10.3390/microorganisms9010186
APA Stylede Vogel, F. A., Schlundt, C., Stote, R. E., Ratto, J. A., & Amaral-Zettler, L. A. (2021). Comparative Genomics of Marine Bacteria from a Historically Defined Plastic Biodegradation Consortium with the Capacity to Biodegrade Polyhydroxyalkanoates. Microorganisms, 9(1), 186. https://doi.org/10.3390/microorganisms9010186