
Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Differentiating Salmonella enterica Serovar Schwarzengrund Isolates Obtained from Food and Animal Sources


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. PFGE
2.3. MLST
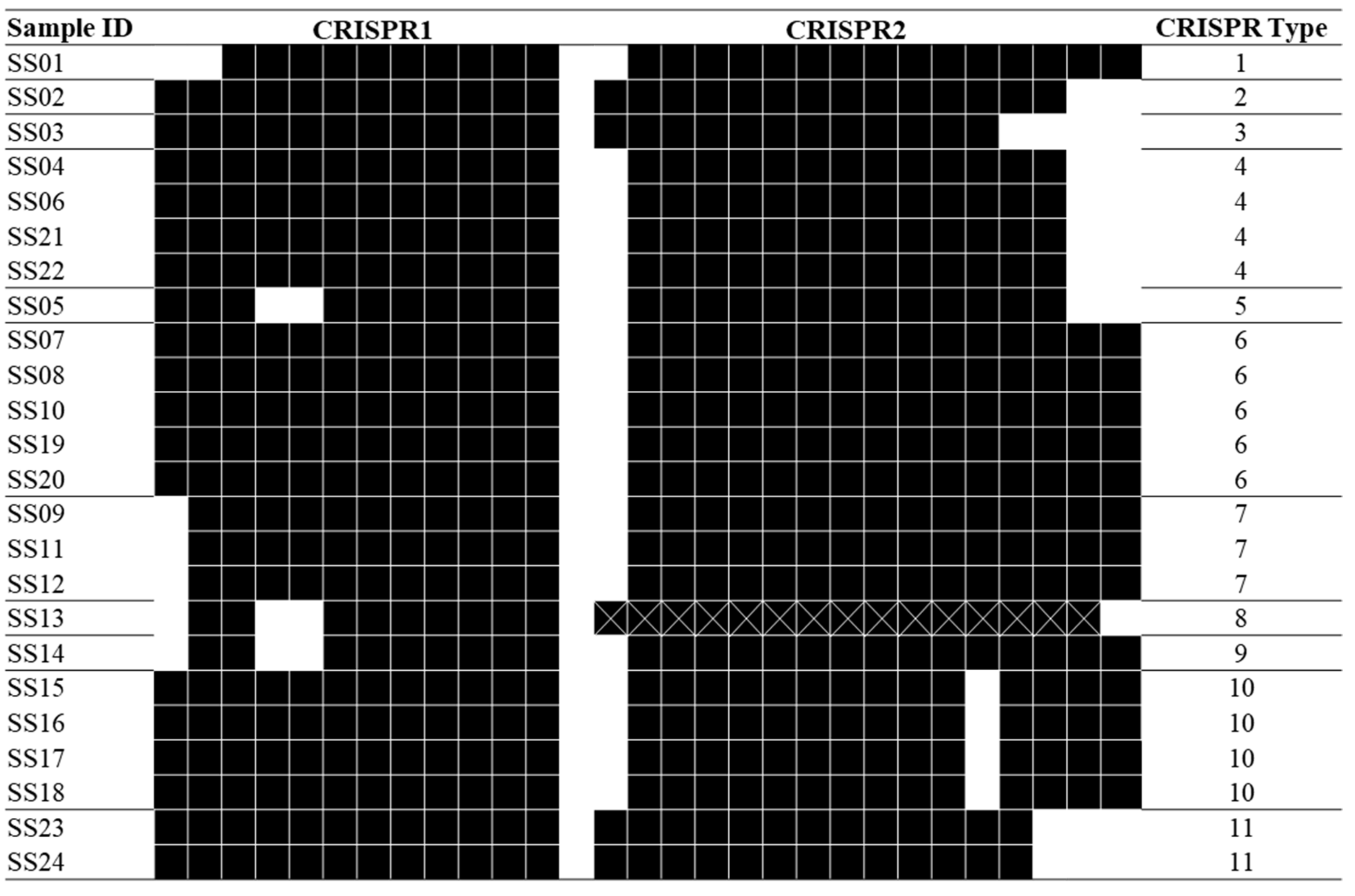
2.4. CRISPR
2.5. WGS
2.6. Data Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Balasubramanian, R.; Im, J.; Lee, J.-S.; Jeon, H.J.; Mogeni, O.D.; Kim, J.H.; Rakotozandrindrainy, R.; Baker, S.; Marks, F. The global burden and epidemiology of invasive non-typhoidal Salmonella infections. Hum. Vaccin. Immunother. 2019, 15, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Ricke, S.C.; Kim, S.A.; Shi, Z.; Park, S.H. Molecular-based identification and detection of Salmonella in food production systems: Current perspectives. J. Appl. Microbiol. 2018, 125, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The Next Culture-Independent Game Changer. Front. Microbiol. 2017, 8, 1069. [Google Scholar] [CrossRef] [Green Version]
- CDC. An Atlas of Salmonella in the United States, 1968–2011: Laboratory-based Enteric Disease Surveillance. Available online: https://www.cdc.gov/salmonella/pdf/salmonella-atlas-508c.pdf (accessed on 15 April 2020).
- Ranieri, M.L.; Shi, C.; Moreno Switt, A.I.; den Bakker, H.C.; Wiedmann, M. Comparison of Typing Methods with a New Procedure Based on Sequence Characterization for Salmonella Serovar Prediction. J. Clin. Microbiol. 2013, 51, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Orsi, R.H.; Luo, H.; Ge, C.; Zhang, G.; Baker, R.C.; Stevenson, A.; Wiedmann, M. Assessment and Comparison of Molecular Subtyping and Characterization Methods for Salmonella. Front. Microbiol. 2019, 10, 1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattiau, P.; Boland, C.; Bertrand, S. Methodologies for Salmonella enterica subsp. enterica subtyping: Gold standards and alternatives. Appl. Environ. Microbiol. 2011, 77, 7877–7885. [Google Scholar] [CrossRef] [Green Version]
- Achtman, M.; Wain, J.; Weill, F.X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.G.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012, 8, e1002776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabre, L.; Zhang, J.; Guigon, G.; Le Hello, S.; Guibert, V.; Accou-Demartin, M.; de Romans, S.; Lim, C.; Roux, C.; Passet, V.; et al. CRISPR Typing and Subtyping for Improved Laboratory Surveillance of Salmonella Infections. PLoS ONE 2012, 7, e36995. [Google Scholar] [CrossRef]
- Sandora, T.J.; Gerner-Smidt, P.; McAdam, A.J. What’s Your Subtype? The Epidemiologic Utility of Bacterial Whole-Genome Sequencing. Clin. Chem. 2020, 60, 586–588. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Tang, H.; Zhao, W.; Meehan, J.; Foley, S.L.; Lin, W.J.; Chen, H.C.; Fang, H.; Nayak, R.; Chen, J.J. Data mining tools for Salmonella characterization: Application to gel-based fingerprinting analysis. BMC Bioinform. 2013, 14 (Suppl. 14), S15. [Google Scholar] [CrossRef] [Green Version]
- Mughini-Gras, L.; Smid, J.; Enserink, R.; Franz, E.; Schouls, L.; Heck, M.; van Pelt, W. Tracing the sources of human salmonellosis: A multi-model comparison of phenotyping and genotyping methods. Infect. Genet. Evol. 2014, 28, 251–260. [Google Scholar] [CrossRef]
- Nadon, C.; Van Walle, I.; Gerner-Smidt, P.; Campos, J.; Chinen, I.; Concepcion-Acevedo, J.; Gilpin, B.; Smith, A.M.; Kam, K.M.; Perez, E.; et al. PulseNet International: Vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Euro. Surveill. 2017, 22, 30544. [Google Scholar] [CrossRef] [PubMed]
- Rumore, J.; Tschetter, L.; Kearney, A.; Kandar, R.; McCormick, R.; Walker, M.; Peterson, C.-L.; Reimer, A.; Nadon, C. Evaluation of whole-genome sequencing for outbreak detection of Verotoxigenic Escherichia coli O157:H7 from the Canadian perspective. BMC Genom. 2018, 19, 870. [Google Scholar] [CrossRef]
- Foley, S.L.; White, D.G.; McDermott, P.F.; Walker, R.D.; Rhodes, B.; Fedorka-Cray, P.J.; Simjee, S.; Zhao, S. Comparison of subtyping methods for differentiating Salmonella enterica serovar Typhimurium isolates obtained from food animal sources. J. Clin. Microbiol. 2006, 44, 3569–3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattaway, M.A.; Dallman, T.J.; Larkin, L.; Nair, S.; McCormick, J.; Mikhail, A.; Hartman, H.; Godbole, G.; Powell, D.; Day, M.; et al. The Transformation of Reference Microbiology Methods and Surveillance for Salmonella With the Use of Whole Genome Sequencing in England and Wales. Front. Public Health 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolar, B.; Joseph, L.A.; Schroeder, M.N.; Stroika, S.; Ribot, E.M.; Hise, K.B.; Gerner-Smidt, P. An Overview of PulseNet USA Databases. Foodborne Pathog. Dis. 2019, 16, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balloux, F.; Brønstad Brynildsrud, O.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Silva, K.C.; Fontes, L.C.; Moreno, A.M.; Astolfi-Ferreira, C.S.; Ferreira, A.J.P.; Lincopan, N. Emergence of extended-spectrum-β-lactamase CTX-M-2-producing Salmonella enterica serovars Schwarzengrund and Agona in poultry farms. Antimicrob. Agents Chemother. 2013, 57, 3458–3459. [Google Scholar] [CrossRef] [Green Version]
- Li, I.C.; Wu, H.H.; Chen, Z.W.; Chou, C.H. Prevalence of IncFIB Plasmids Found among Salmonella enterica Serovar Schwarzengrund Isolates from Animal Sources in Taiwan Using Whole-Genome Sequencing. Pathogens 2021, 10, 1024. [Google Scholar] [CrossRef]
- CDC. Update: Recall of dry dog and cat food products associated with human Salmonella Schwarzengrund infections—United States, 2008. MMWR Morb. Mortal Wkly. Rep. 2008, 57, 1200–1202. [Google Scholar]
- CDC. Outbreak of Salmonella Infections Linked to Butterball Brand Ground Turkey. Available online: https://www.cdc.gov/media/releases/2019/s0315-schwarzengard-salmonella-outbreak.html (accessed on 15 April 2020).
- PAD, G.; FX, W.; Antigenic formulae of the Salmonella serovars, 9th ed. WHO Collaborating Centre for Reference and Research on Salmonella. Available online: https://www.pasteur.fr/sites/default/files/veng_0.pdf (accessed on 15 April 2020).
- PulseNet. Standard Operating Procedure for PulseNet PFGE of Escherichia coli O157:H7, Escherichia coli non-O157 (STEC), Salmonella Serotypes, Shigella sonnei and Shigella flexneri. Available online: http://www.pulsenetinternational.org/assets/PulseNet/uploads/pfge/PNL05_Ec-Sal-ShigPFGEprotocol.pdf (accessed on 15 April 2020).
- Ozdemir, K.; Acar, S. Plasmid Profile and Pulsed–Field Gel Electrophoresis Analysis of Salmonella enterica Isolates from Humans in Turkey. PLoS ONE 2014, 9, e95976. [Google Scholar] [CrossRef] [Green Version]
- Torpdahl, M.; Skov, M.N.; Sandvang, D.; Baggesen, D.L. Genotypic characterization of Salmonella by multilocus sequence typing, pulsed-field gel electrophoresis and amplified fragment length polymorphism. J. Microbiol. Methods 2005, 63, 173–184. [Google Scholar] [CrossRef]
- Liu, F.; Barrangou, R.; Gerner-Smidt, P.; Ribot, E.M.; Knabel, S.J.; Dudley, E.G. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp enterica. Appl. Environ. Microbiol. 2011, 77, 1946–1956. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, P.; Xie, J.; Yi, S.; Yang, C.; Wang, J.; Sun, J.; Liu, N.; Wang, X.; Wu, Z.; et al. New Clustered Regularly Interspaced Short Palindromic Repeat Locus Spacer Pair Typing Method Based on the Newly Incorporated Spacer for Salmonella enterica. J. Clin. Microbiol. 2014, 52, 2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Ang, M.Y.; Dutta, A.; Wee, W.Y.; Dymock, D.; Paterson, I.C.; Choo, S.W. Comparative Genome Analysis of Fusobacterium nucleatum. Genome Biol. Evol. 2016, 8, 2928–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Grundmann, H.; Hori, S.; Tanner, G. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J. Clin. Microbiol. 2001, 39, 4190–4192. [Google Scholar] [CrossRef] [Green Version]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksibi, B.; Ktari, S.; Othman, H.; Ghedira, K.; Maalej, S.; Mnif, B.; Abbassi, M.S.; Fabre, L.; Rhimi, F.; Le Hello, S.; et al. Comparison of conventional molecular and whole-genome sequencing methods for subtyping Salmonella enterica serovar Enteritidis strains from Tunisia. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shariat, N.; Driebe, E.M.; Roe, C.C.; Tolar, B.; Trees, E.; Keim, P.; Zhang, W.; Dudley, E.G.; Fields, P.I.; et al. Comparative analysis of subtyping methods against a whole-genome-sequencing standard for Salmonella enterica serotype Enteritidis. J. Clin. Microbiol. 2015, 53, 212–218. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Murakami, K.; Asai, T.; Etoh, Y.; Ishihara, T.; Kuroki, T.; Horikawa, K.; Fujimoto, S. Multi-locus sequence typing of Salmonella enterica subsp .enterica serovar Enteritidis strains in Japan between 1973 and 2004. Acta Vet. Scand. 2011, 53, 38. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.J.; Lappi, V.; Wolfgang, W.J.; Lapierre, P.; Palumbo, M.J.; Medus, C.; Boxrud, D. Characterization of Foodborne Outbreaks of Salmonella enterica Serovar Enteritidis with Whole-Genome Sequencing Single Nucleotide Polymorphism-Based Analysis for Surveillance and Outbreak Detection. J. Clin. Microbiol. 2015, 53, 3334–3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiden, M.C.J.; Jansen van Rensburg, M.J.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST revisited: The gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Shariat, N.; Dudley, E. CRISPR Typing of Salmonella Isolates. Methods Mol. Biol. 2021, 2182, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Yin, K.; Hu, Y.; Xu, H.; Xie, X.; Xu, L.; Fei, X.; Chen, X.; Jiao, X. Genetic analysis and CRISPR typing of Salmonella enterica serovar Enteritidis from different sources revealed potential transmission from poultry and pig to human. Int. J. Food Microbiol. 2018, 266, 119–125. [Google Scholar] [CrossRef]
- Liu, F.; Kariyawasam, S.; Jayarao, B.M.; Barrangou, R.; Gerner-Smidt, P.; Ribot, E.M.; Knabel, S.J.; Dudley, E.G. Subtyping Salmonella enterica serovar enteritidis isolates from different sources by using sequence typing based on virulence genes and clustered regularly interspaced short palindromic repeats (CRISPRs). Appl. Environ. Microbiol. 2011, 77, 4520–4526. [Google Scholar] [CrossRef] [Green Version]
- Fricke, W.F.; Mammel, M.K.; McDermott, P.F.; Tartera, C.; White, D.G.; Leclerc, J.E.; Ravel, J.; Cebula, T.A. Comparative genomics of 28 Salmonella enterica isolates: Evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 2011, 193, 3556–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touchon, M.; Rocha, E.P.C. The Small, Slow and Specialized CRISPR and Anti-CRISPR of Escherichia and Salmonella. PLoS ONE 2010, 5, e11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neoh, H.M.; Tan, X.E.; Sapri, H.F.; Tan, T.L. Pulsed-field gel electrophoresis (PFGE): A review of the “gold standard” for bacteria typing and current alternatives. Infect. Genet. Evol. 2019, 74, 103935. [Google Scholar] [CrossRef] [PubMed]
- Schürch, A.C.; Arredondo-Alonso, S.; Willems, R.J.L.; Goering, R.V. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene-based approaches. Clin. Microbiol. Infect. 2018, 24, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.E.; Alikhan, N.F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C.J. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar] [CrossRef]
- Mohammed, M.; Thapa, S. Evaluation of WGS-subtyping methods for epidemiological surveillance of foodborne salmonellosis. One Health Outlook 2020, 2, 13. [Google Scholar] [CrossRef]
- Schilbert, H.M.; Rempel, A.; Pucker, B. Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data. Plants 2020, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Boon, E.; Meehan, C.J.; Whidden, C.; Wong, D.H.; Langille, M.G.; Beiko, R.G. Interactions in the microbiome: Communities of organisms and communities of genes. FEMS Microbiol. Rev. 2014, 38, 90–118. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Deloger, M.; El Karoui, M.; Petit, M.-A. A Genomic Distance Based on MUM Indicates Discontinuity between Most Bacterial Species and Genera. J. Bacteriol. 2009, 191, 91. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.L.; Ng, K.P.; Ong, C.S.; Ngeow, Y.F. Genomic Comparisons Reveal Microevolutionary Differences in Mycobacterium abscessus Subspecies. Front. Microbiol. 2017, 8, 2042. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Source | Isolation Year |
|---|---|---|
| SS01 | Duck | 2000 |
| SS02 | Pig | 2000 |
| SS03 | Dog | 2003 |
| SS04 | Pig | 2003 |
| SS05 | Broiler | 2005 |
| SS06 | Pig | 2006 |
| SS07 | Broiler (Farm A; internal control) | 2008 |
| SS08 | Broiler (Farm A) | 2008 |
| SS09 | Broiler (Farm K) | 2008 |
| SS10 | Pet food | 2008 |
| SS11 | Broiler | 2009 |
| SS12 | Broiler | 2010 |
| SS13 | Crested Goshawk | 2011 |
| SS14 | Moorhen | 2011 |
| SS15 | Turkey | 2012 |
| SS16 | Duck | 2012 |
| SS17 | Pig (Farm B) | 2012 |
| SS18 | Pig (Farm C) | 2012 |
| SS19 | Turkey (Farm D) | 2012 |
| SS20 | Turkey (Farm E) | 2012 |
| SS21 | Pig | 2013 |
| SS22 | Broiler | 2014 |
| SS23 | Pig | 2015 |
| SS24 | Broiler | 2018 |
| Method | No. of Types | Discriminatory Power | 95% Confidence Interval |
|---|---|---|---|
| PFGE | 14 | 0.938 | (0.937, 0.939) |
| MLST | 2 | 0.463 | (0.455, 0.471) |
| CRISPR | 11 | 0.906 | (0.905, 0.907) |
| WGS | 20 | 0.982 | (0.982, 0.982) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, I.-C.; Wu, R.; Hu, C.-W.; Wu, K.-M.; Chen, Z.-W.; Chou, C.-H. Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Differentiating Salmonella enterica Serovar Schwarzengrund Isolates Obtained from Food and Animal Sources. Microorganisms 2021, 9, 2046. https://doi.org/10.3390/microorganisms9102046
Li I-C, Wu R, Hu C-W, Wu K-M, Chen Z-W, Chou C-H. Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Differentiating Salmonella enterica Serovar Schwarzengrund Isolates Obtained from Food and Animal Sources. Microorganisms. 2021; 9(10):2046. https://doi.org/10.3390/microorganisms9102046
Chicago/Turabian StyleLi, I-Chen, Rayean Wu, Chung-Wen Hu, Keh-Ming Wu, Zeng-Weng Chen, and Chung-Hsi Chou. 2021. "Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Differentiating Salmonella enterica Serovar Schwarzengrund Isolates Obtained from Food and Animal Sources" Microorganisms 9, no. 10: 2046. https://doi.org/10.3390/microorganisms9102046
APA StyleLi, I.-C., Wu, R., Hu, C.-W., Wu, K.-M., Chen, Z.-W., & Chou, C.-H. (2021). Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Differentiating Salmonella enterica Serovar Schwarzengrund Isolates Obtained from Food and Animal Sources. Microorganisms, 9(10), 2046. https://doi.org/10.3390/microorganisms9102046