
Colonisation Factor CD0873, an Attractive Oral Vaccine Candidate against Clostridioides difficile


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. C. difficile Spore Preparation
2.3. Molecular Manipulations
2.4. Engineering of Antigen Expression Constructs
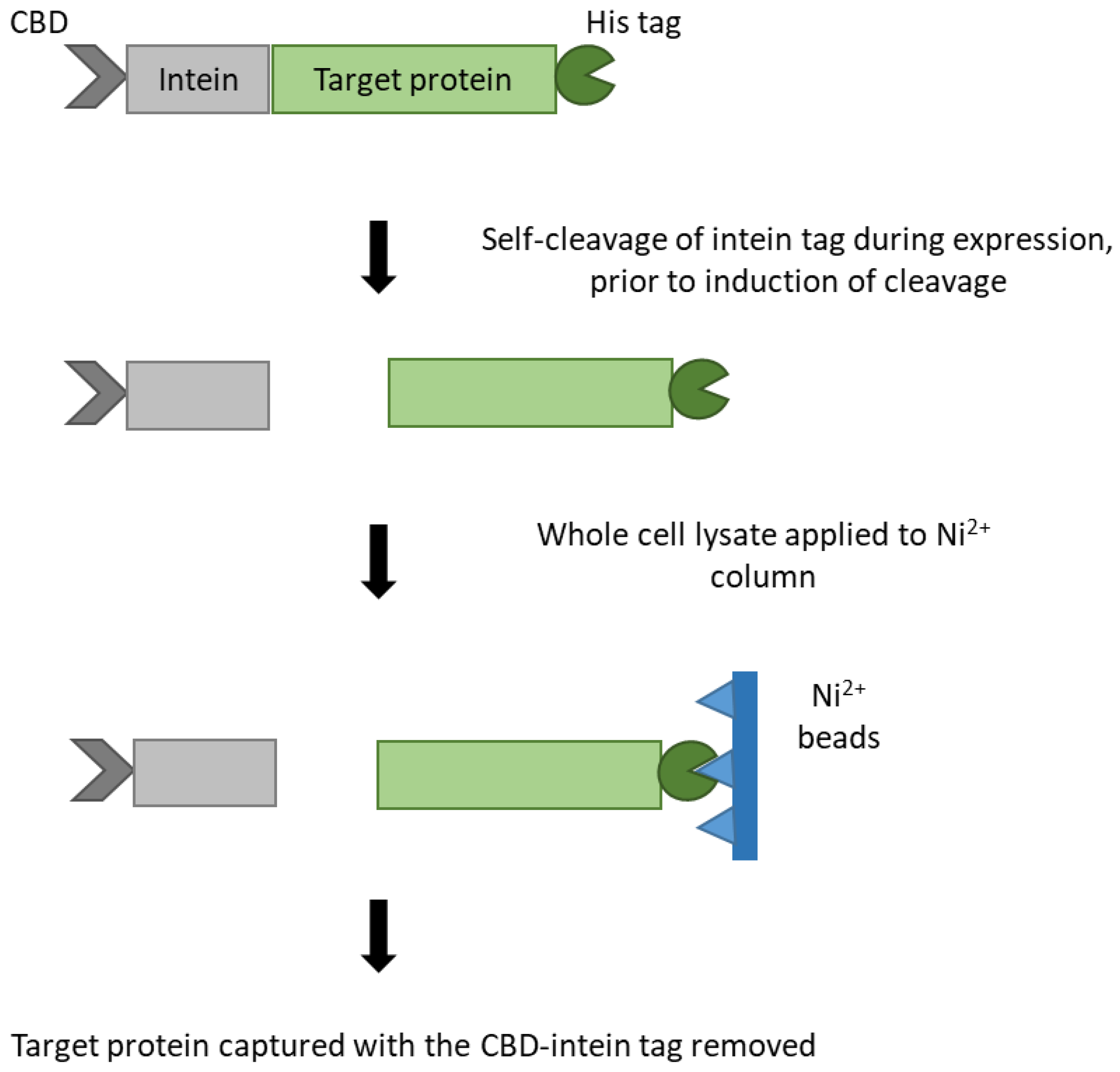
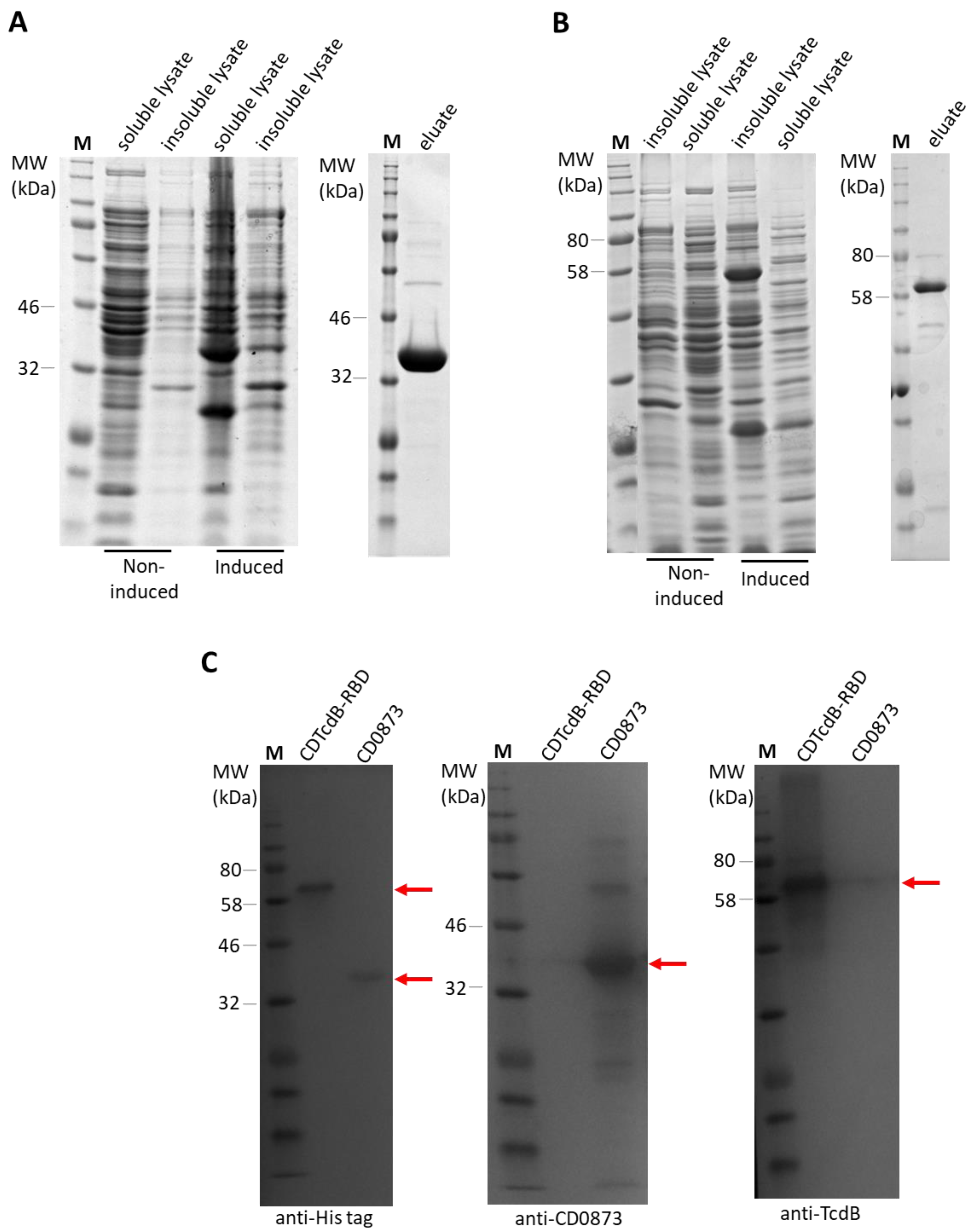
2.5. Expression and Purification of Recombinant Antigens by Immobilised Metal Affinity Chromatography (IMAC)
2.6. Dissolution Assay to Optimise Enteric Coating
2.7. Live CT Imaging to Track Location of Capsule Dissolution
2.8. In Vivo Immunogenicity Study
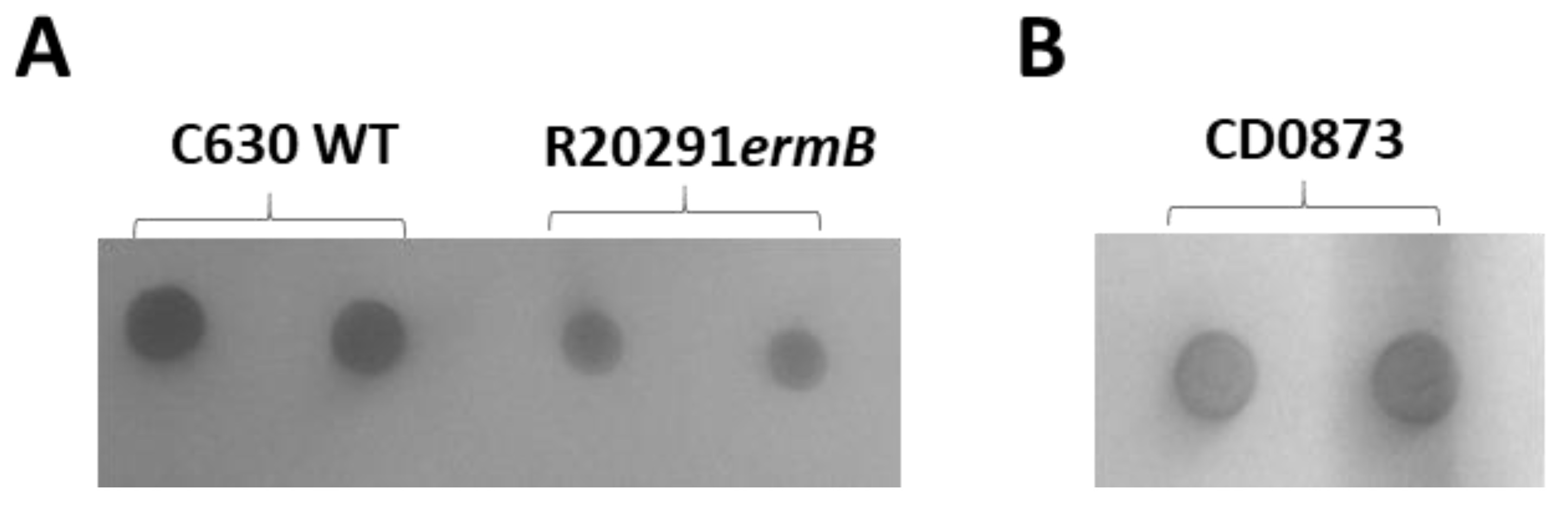
2.9. SDS-PAGE and Western Immunoblotting and Whole Cell Immuno-Dot Blotting
2.10. Indirect ELISA to Assess IgG Levels in Serum
2.11. Toxin Neutralization Assay
2.12. Adherence Blocking Assay
2.13. In Vivo Challenge Study
2.14. Monitoring the Burden of C. difficile
2.15. Histopathological Assessment of Cecum
2.16. Ethics Statement
2.17. Statistical Analysis
3. Results
3.1. Purification of RecombInant Protein Antigens with an N-terminal Cysteine by Novel Double Affinity Tagging
3.2. Single Dip Coating of Gelatin Capsules with EUDRAGIT L100 Mixed with TEC and Water Is Optimal for Targeted Release within the Small Intestine
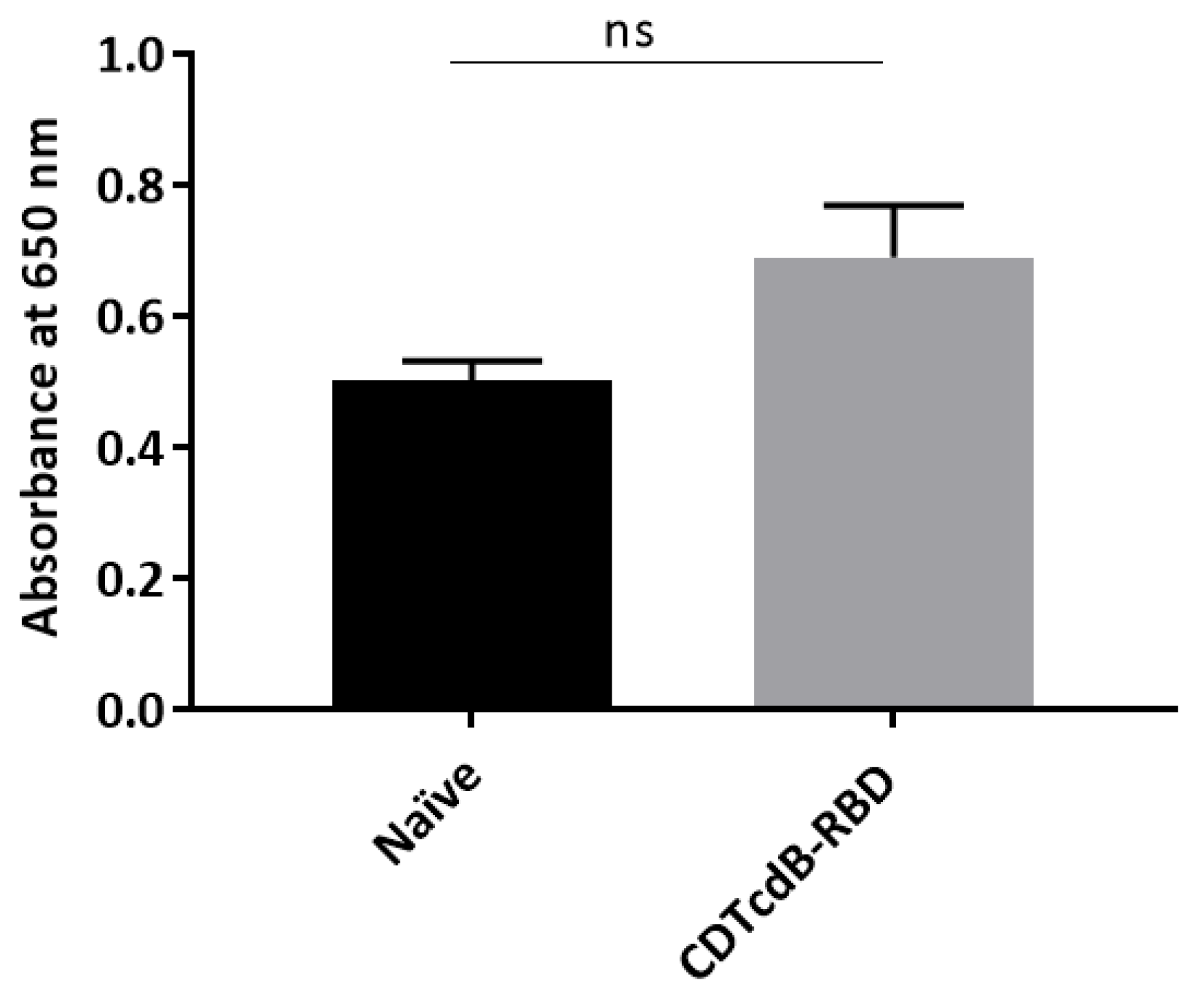
3.3. Mucosal and Systemic Antibody Responses Are Detected in CD0873-Immunised Hamsters
3.4. Intestinal Fluid and Serum of CD0873-Immunised Hamsters Inhibits the Adherence of C. difficile to Caco-2 Cells
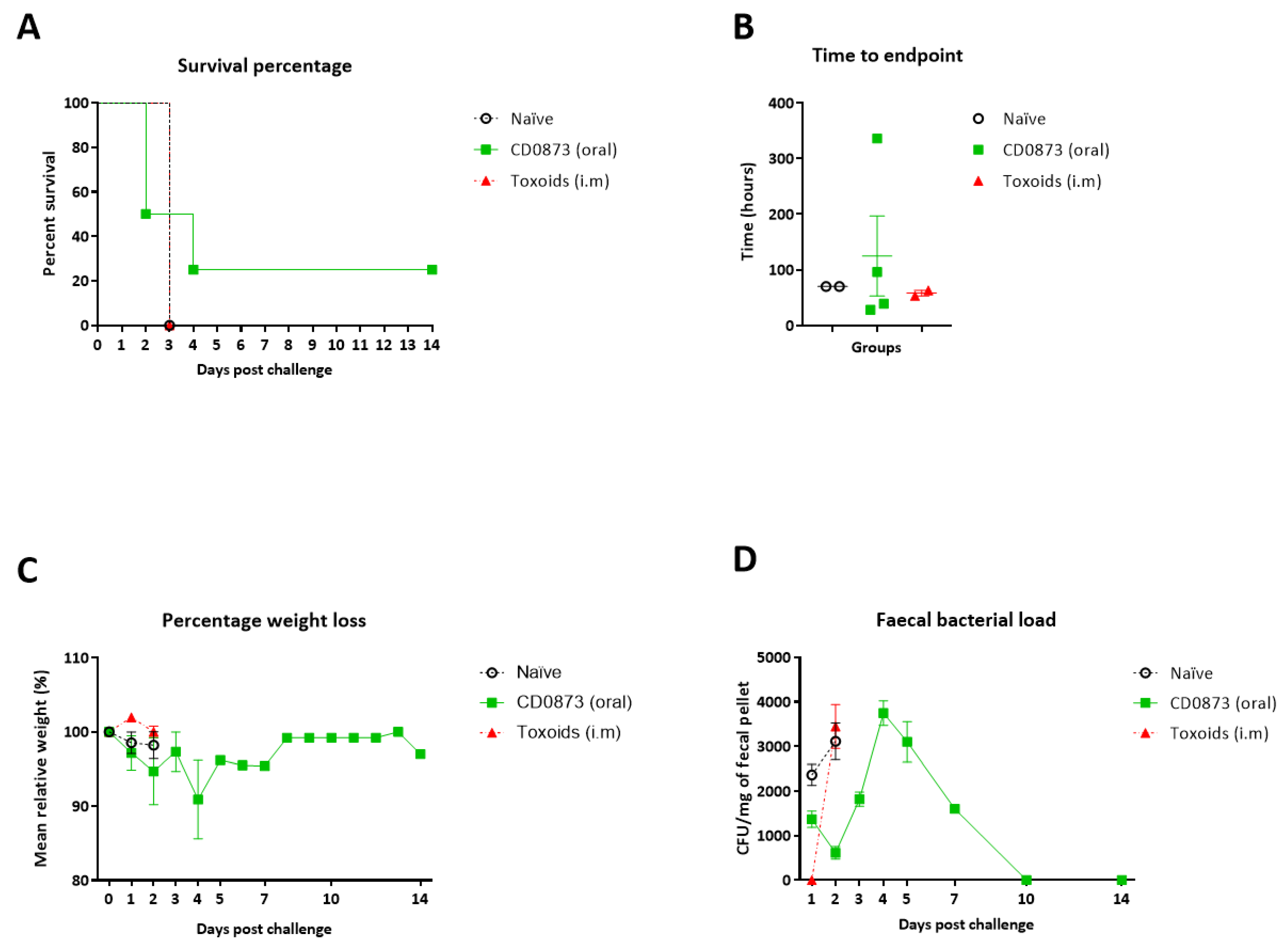
3.5. Hamsters Immunised Orally with CD0873 Are Partially Protected from C. difficile Infection
3.6. Histological Examination of the Cecum Shows Reduced Pathology in CD0873-Immunised Hamsters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Leffler, D.A.; Lamont, J.T. Clostridium difficile infection. N. Engl. J. Med. 2015, 372, 1539–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, T.V.; Lyras, D.; Douce, G.R. Status of vaccine research and development for Clostridium difficile. Vaccine 2019, 37, 7300–7306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Genet. 2009, 7, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.R.; Perencevich, E.N.; Nelson, R.E.; Samore, M.; Khader, K.; Chiang, H.Y.; Chorazy, M.L.; Herwaldt, L.A.; Diekema, D.J.; Kuxhausen, M.F.; et al. Incidence and Outcomes associated with Clostridium difficile infections: A systematic review and meta-analysis. JAMA Netw. Open 2020, 3, e1917597. [Google Scholar] [CrossRef] [Green Version]
- Desai, K.; Gupta, S.B.; Dubberke, E.R.; Prabhu, V.S.; Browne, C.; Mast, T.C. Epidemiological and economic burden of Clostridium difficile in the United States: Estimates from a modeling approach. BMC Infect. Dis. 2016, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hensgens, M.P.; Goorhuis, A.; Dekkers, O.M.; Kuijper, E.J. Time interval of increased risk for Clostridium difficile infection after exposure to antibiotics. J. Antimicrob. Chemother. 2011, 67, 742–748. [Google Scholar] [CrossRef]
- Hung, Y.-P.; Lin, H.-J.; Wu, T.-C.; Liu, H.-C.; Lee, J.-C.; Lee, C.-I.; Wu, Y.-H.; Wan, L.; Tsai, P.-J.; Ko, W.-C. Risk factors of fecal toxigenic or non-toxigenic Clostridium difficile colonization: Impact of toll-like receptor polymorphisms and prior antibiotic exposure. PLoS ONE 2013, 8, e69577. [Google Scholar] [CrossRef]
- Loo, V.G.; Bourgault, A.M.; Poirier, L.; Lamothe, F.; Michaud, S.; Turgeon, N.; Toye, B.; Beaudoin, A.; Frost, E.H.; Gilca, R.; et al. Host and pathogen factors for Clostridium difficile infection and colonization. N. Engl. J. Med. 2011, 365, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Cule, M.L.; Wilson, D.J.; Griffiths, D.; Vaughan, A.; O’Connor, L.; Ip, C.L.C.; Golubchik, T.; Batty, E.M.; Finney, J.M.; et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N. Engl. J. Med. 2013, 369, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Clabots, C.R.; Johnson, S.; Olson, M.M.; Peterson, L.R.; Gerding, D.N. Acquisition of clostridium difficile by hospitalized patients: Evidence for colonized new admissions as a source of infection. J. Infect. Dis. 1992, 166, 561–567. [Google Scholar] [CrossRef]
- Marciniak, C.; Chen, D.; Stein, A.C.; Semik, P.E. Prevalence of Clostridium difficile colonization at admission to rehabilitation. Arch. Phys. Med. Rehabil. 2006, 87, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Gerding, D.N.; Olson, M.M.; Weiler, M.D.; Hughes, R.A.; Clabots, C.R.; Peterson, L.R. Prospective, controlled study of vinyl glove use to interrupt Clostridium difficile nosocomial transmission. Am. J. Med. 1990, 88, 137–140. [Google Scholar] [CrossRef]
- Hung, Y.-P.; Lee, J.; Lin, H.-J.; Liu, H.-C.; Wu, Y.-H.; Tsai, P.-J.; Ko, W.-C. Clinical impact of Clostridium difficile colonization. J. Microbiol. Immunol. Infect. 2015, 48, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Pardi, D.S.; Aronson, S.L.; Kammer, P.P.; Orenstein, R.; St Sauver, J.L.; Harmsen, W.S.; Zinsmeister, A.R. The Epidemiology of community-acquired Clostridium difficile infection: A population-based study. Am. J. Gastroenterol. 2012, 107, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochan, T.J.; Somers, M.J.; Kaiser, A.M.; Shoshiev, M.S.; Hagan, A.K.; Hastie, J.L.; Giordano, N.P.; Smith, A.D.; Schubert, A.M.; Carlson, P.E.; et al. Intestinal calcium and bile salts facilitate germination of Clostridium difficile spores. PLoS Pathog. 2017, 13, e1006443. [Google Scholar] [CrossRef]
- Denève, C.; Janoir, C.; Poilane, I.; Fantinato, C.; Collignon, A. New trends in Clostridium difficile virulence and pathogenesis. Int. J. Antimicrob. Agents 2009, 33, S24–S28. [Google Scholar] [CrossRef]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; Di Masi, A. Clostridium difficile Toxins A and B: Insights into pathogenic properties and extraintestinal effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [Green Version]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Infect. Dis. Soc. Am. 2018, 66, e1–e48. [Google Scholar]
- Czepiel, J.; Kędzierska, J.; Biesiada, G.; Birczyńska, M.; Perucki, W.; Nowak, P.; Garlicki, A. Epidemiology of Clostridium difficile infection: Results of a hospital-based study in Krakow, Poland. Epidemiol. Infect. 2015, 143, 3235–3243. [Google Scholar] [CrossRef]
- Cohen, S.H.; Gerding, D.N.; Johnson, S.; Kelly, C.P.; Loo, V.G.; McDonald, L.C.; Pepin, J.; Wilcox, M.H. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect. Control. Hosp. Epidemiol. 2010, 31, 431–455. [Google Scholar] [CrossRef]
- Debast, S.; Bauer, M.P.; Kuijper, E.J. European society of clinical microbiology and infectious diseases: Update of the treatment guidance document for Clostridium difficile infection. Clin. Microbiol. Infect. 2014, 20, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moura, H.; Terilli, R.R.; Woolfitt, A.R.; Williamson, Y.M.; Wagner, G.; Blake, T.A.; Solano, M.I.; Barr, J.R. Proteomic analysis and label-free quantification of the large Clostridium difficile toxins. Int. J. Proteom. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, L.V.; Elmer, G.W.; Surawicz, C.M. Breaking the cycle: Treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am. J. Gastroenterol. 2002, 97, 1769–1775. [Google Scholar] [CrossRef]
- Dutta, S.K.; Girotra, M.; Garg, S.; Dutta, A.; Von Rosenvinge, E.C.; Maddox, C.; Song, Y.; Bartlett, J.G.; Vinayek, R.; Fricke, W.F. Efficacy of combined jejunal and colonic fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin. Gastroenterol. Hepatol. 2014, 12, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.; Cannon, K.; O’grady, H.; Wu, K.; Ward, L. Fecal microbiome transplantation (FMT) via oral fecal microbial capsules for recurrent Clostridium difficile infection (rCDI). In Proceedings of the IDWeek 2013 Meeting of the Infectious Diseases Society of America, San Francisco, CA, USA, 2–6 October 2013; p. 201389. [Google Scholar]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile toxin biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef]
- Katchar, K.; Taylor, C.P.; Tummala, S.; Chen, X.; Sheikh, J.; Kelly, C.P. Association Between IgG2 and IgG3 Subclass responses to toxin A and recurrent Clostridium difficile–associated disease. Clin. Gastroenterol. Hepatol. 2007, 5, 707–713. [Google Scholar] [CrossRef]
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C.; et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med. 2017, 376, 305–317. [Google Scholar] [CrossRef]
- Wang, J.; Thorson, L.; Stokes, R.W.; Santosuosso, M.; Huygen, K.; Zganiacz, A.; Hitt, M.; Xing, Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004, 173, 6357–6365. [Google Scholar] [CrossRef]
- Vela Ramirez, J.E.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef]
- Azizi, A.; Kumar, A.; Diaz-Mitoma, F.; Mestecky, J. Enhancing oral vaccine potency by targeting intestinal M cells. PLOS Pathog. 2010, 6, e1001147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsted, D.; Fallahi, F.; Golshani, A.; Azizi, A. Advances and challenges in mucosal adjuvant technology. Vaccine 2015, 33, 2399–2405. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, H.; Zhang, X.; Qian, F. Intranasal and oral vaccination with protein-based antigens: Advantages, challenges and formulation strategies. Protein Cell 2015, 6, 480–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, E.L.; Freeman, J.; Wilcox, M.H. Models for the study of Clostridium difficile infection. Gut Microbes 2012, 3, 145–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.; Drudy, D.; Kyne, L.; Brown, K.; Fairweather, N.F. Immunoreactive cell wall proteins of Clostridium difficile iden-tified by human sera. J. Med. Microbiol. 2008, 57, 750–756. [Google Scholar] [CrossRef]
- Bradshaw, W.J.; Bruxelle, J.F.; Kovacs-Simon, A.; Harmer, N.J.; Janoir, C.; Péchiné, S.; Acharya, K.R.; Michell, S.L. Molecular features of lipoprotein CD0873: A potential vaccine against the human pathogen Clostridioides difficile. J. Biol. Chem. 2019, 294, 15850–15861. [Google Scholar] [CrossRef] [Green Version]
- Kovacs-Simon, A.; Leuzzi, R.; Kasendra, M.; Minton, N.P.; Titball, R.W.; Michell, S.L. Lipoprotein CD0873 is a novel adhesin of Clostridium difficile. J. Infect. Dis. 2014, 210, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Belyi, I.F.; Varfolomeeva, N.A. Construction of a fusion protein carrying antigenic determinants of enteric clostridial toxins. FEMS Microbiol. Lett. 2003, 225, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Pavliakova, D.; Moncrief, J.S.; Lyerly, D.M.; Schiffman, G.; Bryla, D.A.; Robbins, J.B.; Schneerson, R. Clostridium difficile recombinant toxin a repeating units as a carrier protein for conjugate vaccines: Studies of pneumococcal Type 14, Escherichia coli K1, and Shigella flexneri Type 2a polysaccharides in mice. Infect. Immun. 2000, 68, 2161–2166. [Google Scholar] [CrossRef] [Green Version]
- Sauerborn, M.; Leukel, P.; von Eichel-Streiber, C. The C-terminal ligand-binding domain of Clostridium difficile toxin A (TcdA) abrogates TcdA-specific binding to cells and prevents mouse lethality. FEMS Microbiol. Lett. 1997, 155, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wren, B.W.; Russell, R.R.; Tabaqchali, S. Antigenic cross-reactivity and functional inhibition by antibodies to Clostridium difficile toxin A, Streptococcus mutans glucan-binding protein, and a synthetic peptide. Infect. Immun. 1991, 59, 3151–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyerly, D.M.; Johnson, J.L.; Frey, S.M.; Wilkins, T.D. Vaccination against lethal Clostridium difficile enterocolitis with a nontoxic recombinant peptide of toxin A. Curr. Microbiol. 1990, 21, 29–32. [Google Scholar] [CrossRef]
- Kink, J.A.; Williams, J.A. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 1998, 66, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.L.; Ng, Y.K.; Cartman, S.T.; Collery, M.M.; Cockayne, A.; Minton, N.P. Improving the reproducibility of the NAP1/B1/027 epidemic strain R20291 in the hamster model of infection. Anaerobe 2016, 39, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Staelens, D.; Liang, S.; Appeltans, B.; Van de Wouwer, M.; Van den Mooter, G.; Van Assche, G.; Himmelreich, U.; Vande Velde, G. Visualization of delayed release of compounds from pH-sensitive capsules in vitro and in vivo in a hamster model. Contrast Med. Mol. Imaging 2016, 11, 24–31. [Google Scholar] [CrossRef]
- Da Silva, R.A.G.; Churchward, C.P.; Karlyshev, A.V.; Eleftheriadou, O.; Snabaitis, A.K.; Longman, M.R.; Ryan, A.; Griffin, R. The role of apolipoprotein N-acyl transferase, Lnt, in the lipidation of factor H binding protein of Neisseria meningitidis strain MC58 and its potential as a drug target. Br. J. Pharmacol. 2017, 174, 2247–2260. [Google Scholar] [CrossRef] [Green Version]
- Anosova, N.G.; Brown, A.M.; Li, L.; Liu, N.; Cole, L.E.; Zhang, J.; Mehta, H.; Kleanthous, H. Systemic antibody responses induced by a two-component Clostridium difficile toxoid vaccine protect against C. difficile-associated disease in hamsters. J. Med. Microbiol. 2013, 62, 1394–1404. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Collery, M.M.; Kelly, M.L.; Cartman, S.T.; Cockayne, A.; Minton, N.P. Importance of Toxin A, Toxin B, and CDT in virulence of an epidemic Clostridium difficile Strain. J. Infect. Dis. 2014, 209, 83–86. [Google Scholar] [CrossRef]
- Pawlowski, S.W.; Calabrese, G.; Kolling, G.L.; Platts-Mills, J.; Freire, R.; AlcantaraWarren, C.; Liu, B.; Sartor, R.B.; Guerrant, R.L. Murine model of Clostridium difficile infection with aged gnotobiotic C57BL/6 Mice and a BI/NAP1 Strain. J. Infect. Dis. 2010, 202, 1708–1712. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The arrive guidelines for reporting animal research. J. Pharmacol. Pharmacother. 2010, 1, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leher, H.F.; Alizadeh, H.; Taylor, W.M.; Shea, A.S.; Silvany, R.S.; Van Klink, F.; Jager, M.J.; Niederkorn, J.Y. Role of mucosal IgA in the resistance to Acanthamoeba keratitis. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2666–2673. [Google Scholar]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandtzaeg, P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine 2007, 25, 5467–5484. [Google Scholar] [CrossRef]
- MacPherson, A.J.; McCoy, K.D.; Johansen, F.-E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 2007, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Czerkinsky, C.; Holmgren, J. Vaccines against enteric infections for the developing world. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150142. [Google Scholar] [CrossRef] [Green Version]
- Aboudola, S.; Kotloff, K.L.; Kyne, L.; Warny, M.; Kelly, E.C.; Sougioultzis, S.; Giannasca, P.J.; Monath, T.P.; Kelly, C.P. Clostridium difficile vaccine and serum immunoglobulin G antibody response to toxin, A. Infect. Immun. 2003, 71, 1608–1610. [Google Scholar] [CrossRef] [Green Version]
- Aronsson, B.; Granström, M.; Möllby, R.; Nord, C.E. Serum antibody response to Clostridium difficile toxins in patients with Clostridium difficile diarrhoea. Infection 1985, 13, 97–101. [Google Scholar] [CrossRef]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001, 357, 189–193. [Google Scholar] [CrossRef]
- Giannasca, P.J.; Zhang, Z.X.; Lei, W.D.; Boden, J.A.; Giel, M.A.; Monath, T.P.; Thomas, W.D., Jr. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 1999, 67, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.H.; Iaconis, J.P.; Rolfe, R.D. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect. Immun. 1987, 55, 2984–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.F.; Lyerly, D.M.; Hill, J.E.; Monath, T.P. Evaluation of formalin-inactivated Clostridium difficile vaccines administered by parenteral and mucosal routes of immunization in hamsters. Infect. Immun. 1995, 63, 4619–4627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, X.; Zhang, Y.; Li, S.; Chen, K.; Shi, L.; Nie, W.; Kumar, R.; Tzipori, S.; Wang, J.; et al. A Chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect. Immun. 2012, 80, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.H.; Fuhrmann, S.R.; Kluepfel-Stahl, S.; Carman, R.J.; Ellingsworth, L.; Flyer, D.C. A novel fusion protein containing the receptor binding domains of C. difficile toxin A and toxin B elicits protective immunity against lethal toxin and spore challenge in preclinical efficacy models. Vaccine 2012, 30, 4249–4258. [Google Scholar] [CrossRef]
- Siddiqui, F.; O’Connor, J.R.; Nagaro, K.; Cheknis, A.; Sambol, S.P.; Vedantam, G.; Gerding, D.N.; Johnson, S. Vaccination with parenteral toxoid B protects hamsters against lethal challenge with toxin A–negative, toxin B–positive Clostridium difficile but does not prevent colonization. J. Infect. Dis. 2011, 205, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.H.; Hong, S.J.; Lee, Y.-K.; Cho, S. Oral Vaccine delivery for intestinal immunity—Biological basis, barriers, delivery system, and m cell targeting. Polymers 2018, 10, 948. [Google Scholar] [CrossRef] [Green Version]
- Kaul, D.; Ogra, P.L. Mucosal responses to parenteral and mucosal vaccines. Dev. Boil. Stand. 1998, 95, 141–146. [Google Scholar]
- Cartman, S.T.; Heap, J.T.; Kuehne, S.A.; Cockayne, A.; Minton, N.P. The emergence of ‘hypervirulence’ in Clostridium difficile. Int. J. Med. Microbiol. 2010, 300, 387–395. [Google Scholar] [CrossRef]
- Hong, H.A.; Hitri, K.; Hosseini, S.; Kotowicz, N.; Bryan, D.; Mawas, F.; Wilkinson, A.J.; van Broekhoven, A.; Kearsey, J.; Cutting, S.M. Mucosal antibodies to the C terminus of toxin A prevent colonization of Clostridium difficile. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [Green Version]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef]
- Apter, F.M.; Michetti, P.; Winner, L.D.; Mack, J.A.; Mekalanos, J.J.; Neutra, M.R. Analysis of the roles of antilipopoly-saccharide and anti-cholera toxin immunoglobulin A (IgA) antibodies in protection against Vibrio cholerae and cholera toxin by use of monoclonal IgA antibodies in vivo. Infect. Immun. 1993, 61, 5279–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, J.; Czerkinsky, C.; Lycke, N.; Svennerholm, A.-M. Mucosal immunity: Implications for vaccine development. Immunobiology 1992, 184, 157–179. [Google Scholar] [CrossRef]
- Dallas, S.D.; Rolfe, R.D. Binding of Clostridium difficile toxin A to human milk secretory component. J. Med. Microbiol. 1998, 47, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permpoonpattana, P.; Hong, H.A.; Phetcharaburanin, J.; Huang, J.M.; Cook, J.; Fairweather, N.F.; Cutting, S.M. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect. Immun. 2011, 79, 2295–2302. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karyal, C.; Hughes, J.; Kelly, M.L.; Luckett, J.C.; Kaye, P.V.; Cockayne, A.; Minton, N.P.; Griffin, R. Colonisation Factor CD0873, an Attractive Oral Vaccine Candidate against Clostridioides difficile. Microorganisms 2021, 9, 306. https://doi.org/10.3390/microorganisms9020306
Karyal C, Hughes J, Kelly ML, Luckett JC, Kaye PV, Cockayne A, Minton NP, Griffin R. Colonisation Factor CD0873, an Attractive Oral Vaccine Candidate against Clostridioides difficile. Microorganisms. 2021; 9(2):306. https://doi.org/10.3390/microorganisms9020306
Chicago/Turabian StyleKaryal, Cansu, Jaime Hughes, Michelle L. Kelly, Jeni C. Luckett, Philip V. Kaye, Alan Cockayne, Nigel P. Minton, and Ruth Griffin. 2021. "Colonisation Factor CD0873, an Attractive Oral Vaccine Candidate against Clostridioides difficile" Microorganisms 9, no. 2: 306. https://doi.org/10.3390/microorganisms9020306