Abstract
DNA polymerase B1 (PolB1) is a member of the B-family DNA polymerase family and is a replicative DNA polymerase in Crenarchaea. PolB1 is responsible for the DNA replication of both the leading and lagging strands in the thermophilic crenarchaeon Sulfolobus acidocaldarius. Recently, two subunits, PolB1-binding protein (PBP)1 and PBP2, were identified in Saccharolobus solfataricus. Previous in vitro studies suggested that PBP1 and PBP2 influence the core activity of apoenzyme PolB1 (apo-PolB1). PBP1 contains a C-terminal acidic tail and modulates the strand-displacement synthesis activity of PolB1 during the synthesis of Okazaki fragments. PBP2 modestly enhances the DNA polymerase activity of apo-PolB1. These subunits are present in Sulfolobales, Acidilobales, and Desulfurococcales, which belong to Crenarchaea. However, it has not been determined whether these subunits are essential for the activity of apo-PolB1. In this study, we constructed a pbp1 deletion strain in S. acidocaldarius and characterized its phenotypes. However, a pbp2 deletion strain was not obtained, indicating that PBP2 is essential for replication by holoenzyme PolB1. A pbp1 deletion strain was sensitive to various types of DNA damage and exhibited an increased mutation rate, suggesting that PBP1 contribute to the repair or tolerance of DNA damage by holoenzyme PolB1. The results of our study suggest that PBP1 is important for DNA repair by holoenzyme PolB1 in S. acidocaldarius.
1. Introduction
DNA polymerases (DNAPs) are enzymes that synthesize DNA, playing a central role in DNA replication and repair. Accurate and timely replication is important for all living organisms. In general, replicative DNAPs are highly processive, accurate, and exhibit 3′ to 5′ exonuclease activity [1]. DNA damage is largely unavoidable, and efficient repair of that is important for accurate DNA replication [2,3]. Generally, non-replicative DNAPs are responsible for various and often short-length DNA synthesis in repair. In bacteria, a C-family polymerase, namely, PolIII, synthesizes the leading and lagging strands. PolIII consists of a ten-component complex: The catalytic part (α-, ε-, and θ-subunits), the clamp loader or γ-complex (γ-, δ-, δ’-, ζ-, χ-, and ψ-subunits), and the sliding clamp (β2) [4,5]. In Eukarya, two B-family polymerases, Polε and Polδ, replicate the leading and lagging strands, respectively [6,7]. These DNAPs are multi-subunit proteins containing a catalytic subunit, a regulatory subunit, and an assortment of accessory subunits [5,8,9]. Most archaea except for Crenarchaea possess a D-family polymerase and at least one B-family polymerase [7,10,11]. The B-family polymerase PolB3 is distributed widely in almost all archaea except Thaumarchaota [10,11]. The euryarchaea Methanococcus maripaludis and Thermococcus kodakarensis have PolD and PolB3. In genetic studies of these species, polD is essential for viability, but polB3 is not; that is, PolD replicates both the leading and lagging strands [12,13,14]. Crenarchaea lack PolD, but possess at least two B-family polymerases [7,10,11]. The extremely thermophilic crenarchaeon Sulfolobus acidocaldarius has four DNAPs: PolB1, PolB2, PolB3, and Dbh. Previous in vivo experiments indicated that PolB1 is a replicative polymerase for both leading and lagging strands since the triple gene-deletion strains lacking polB2, polB3, and dbh had been successfully isolated [15]. In short, it is plausible that PolD in Euryarchaea and PolB1 in Crenarchaea are replicative DNAPs [11,12,14,15,16].
PolD is composed of a large catalytic subunit (DP2) and a smaller subunit with 3′ to 5′ exonuclease activity (DP1) [17,18]. On the other hand, PolB1 has been believed to be a single-subunit enzyme since the characterization of PolB1 in Sulfolobus acidocaldarius in 1985 [19]. In 2017, two subunits, PolB1-binding protein (PBP)1 and PBP2, were identified in Saccharolobus solfataricus [20,21]. PolB1 was revealed to be a multi-subunit protein. PBP1 and PBP2 influence the core activity of apoenzyme PolB1 (apo-PolB1) [20]. PBP1 contains a C-terminal acidic tail and modulates the strand-displacement synthesis activity of PolB1 during the synthesis of Okazaki fragments [20]. Thus, PBP1 limits the needless elimination and resynthesis of DNA in the preceding Okazaki fragment for efficient lagging strand DNA synthesis [20]. PBP2 modestly increases the DNA polymerase activity of apo-PolB1 [20,22]. In addition, it reduces the inhibition of DNA synthesis by PBP1 [20,22]. These subunits are present in Sulfolobales, Acidilobales, and Desulfurococcales, which belong to Crenarchaea. However, it has not been determined whether these subunits are essential for the activity of apo-PolB1.
To examine whether these subunits are essential for the activity of apo-PolB1 in S. acidocaldarius, we attempted to construct strains completely lacking the pbp1 and pbp2 genes and characterized their mutant phenotypes, examining sensitivity to numerous types of DNA damage (i.e., UV irradiation, DNA-damaging agents, heat shock, and DNA replication inhibitors) and mutation rates. We report that holoenzyme PolB1 (apo-PolB1 with PBP1 and PBP2) is responsible for the repair of most DNA damage in addition to DNA replication in S. acidocaldarius.
2. Materials and Methods
2.1. Strains and Growth Conditions
The growth conditions were previously reported [23]. S. acidocaldarius strain DP-1 (ΔpyrE ΔsuaI Δphr), which is pyrimidine-auxotrophic, restriction endonuclease SuaI-deficient and DNA photolyase Phr-deficient was used as the parent strain [23,24] for construction of strain HM-8 (Table 1). These strains were cultivated in the xylose and tryptone (XT) medium (pH 3) [15,25] at 75 °C with or without shaking (160 rpm). For plate medium, identical components of 1× basal salts containing 2.9 g MgSO4·7 H2O and 0.5 g CaCl2·2H2O, and Gellan Gum (0.65 g/L) were used. Uracil (0.02 g/L) was added to XT medium (XTU) for cultivation of pyrimidine-auxotrophic strain. 5-fluoro-orotic acid (FOA) (50 μg/mL) was added to the XTU medium (XTUF) for counter selection in the pop-out recombination (Section 2.3) and for spontaneous mutation analysis (Section 2.7).

Table 1.
Strains and DNA sequences used in this study.
2.2. General DNA Manipulation
The reagents used in these experiments were prepared as previously described [23]. EmeraldAmp MAX PCR Master Mix (Takara Bio, Kusatsu, Shiga, Japan) was used for PCR amplification. PCR products were purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany). The Sanger sequencing was performed by the Eurofins Genomics (Tokyo, Japan, https://www.eurofinsgenomics.jp/).
2.3. Construction of the PolB1-binding Protein Gene-Deleted Strains
The multiple gene knockout strategy with one-step PCR (MONSTER) was used to prepare pbp1 (Saci_0746) and pbp2 (Saci_1566) knockout cassettes (MONSTER-pbp1 and MONSTER-pbp2, respectively) and to construct pbp1 and pbp2 deletion strains [23]. In addition, another pbp2 knockout cassette (MONSTER-pbp2n) was prepared to delete pbp2 in different deletion regions. The DNAs and PCR primers used in this study are listed in Table 1 and Table 2, respectively. In brief, the MONSTER-pbp1 cassette was amplified from placSpyrE as a template using the MONSTER-pbp1-F/R primers. Similarly, the MONSTER-pbp2 and MONSTER-pbp2n cassettes were amplified using MONSTER-pbp2-F/R primers and MONSTER-pbp2n-F/R primers, respectively. The purified PCR products (100–200 ng/μL in 5 mM Tris-HCl, pH 8.5) were used for subsequent electrotransformation.
Table 2.
Primers used in this study.
The transformation procedure has been previously described in detail [23]. To delete pbp1, 2 μg of MONSTER-pbp1 was electroporated (15 kV/cm, 9 ms) into 200 μL of DP-1 competent cells harvested at the mid-log phase (the optical density of the culture at 600 nm (OD600] = 0.34–0.43) in a 2 mm electroporation cuvette (Bio-Rad, Hercules, CA, USA). Similarly, MONSTER-pbp2 or MONSTER-pbp2n was electroporated into 200 μL of DP-1 competent cells to delete pbp2. After electroporation, the sample was spread onto an XT plate. After five days of cultivation at 75 °C, blue transformant colonies were selected by spraying a 10 mg/mL 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) solution in 40% N,N-dimethylformamide (DMF) diluted with 0.85% NaCl solution on the plate, followed by incubation at 75 °C for one day. The genotypes were confirmed using the outer primers (pbp1-out-F/R). Single-colony isolation followed by genotypic analysis using the outer primers was performed at each step for the selection of intermediates and gene deletion strains. To exclude translocation of the pbp1 gene in any genomic locus of the pbp1-deleted strain HM-8, PCR analysis was performed using inner primers pbp1-in-F/R, which anneal with the inner (deleted) region of the pbp1 gene.
The deletion of the pbp1 gene was also checked by sequencing analysis. A pbp1 gene was amplified from cultures of DP-1 and HM-8 using the outer primers (pbp1-out-F/R). Each pbp1 gene was sequenced by Sanger method using the outer primer (pbp1-out-F) (Table 2).
2.4. Construction of the pyrE-Proficient Strains
The pyrE-proficient strain was constructed as previously described [26]. A short cassette carrying 18 bp-deletion of pyrE gene sequence of the pyrimidine-auxotrophic strain MR31 [27], and 150 bp and 101 bp of the 5′ and 3′ flanking regions, respectively, was amplified from the S. acidocaldarius DSM639 genomic DNA using SAMR31-F/R primers under the following conditions: 94 °C for 3 min; 30 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 3 min. The purified PCR products were electroporated (15 kV/cm, 9 ms) into 200 μL of competent cells of the pyrimidine-auxotrophic strains DP-1 or HM-8 harvested at early to mid-log phase (OD600 = 0.34 and 0.30, respectively), and the resulting colonies were isolated. After a second single-colony isolation, the manipulated regions of genomic DNA of each strain were checked by PCR using SAMR31-F/R primers. The strain containing the expected lengthening of this interval was used as the pyrE-proficient strain.
2.5. Growth Temperature Range
For characterization of the growth temperature range, overnight cultures of DP-1 and HM-8 (late-log to stationary phase) were inoculated into 6 mL of XTU liquid medium to yield an initial OD600 = 0.005 in triplicate. The cells in loosely capped glass tubes were cultivated at 50–80 °C (temperature range from minimal to maximal growth temperature) with intervals of 5 °C without shaking on the block heater.
2.6. DNA Damage Sensitivity Tests
The sensitivity tests to the UV irradiation or DNA-damaging agents of the mutant and parental strains were performed by using the exact same protocol as previously described [23]. The survivability test after exposure of DNA damaging agents was also performed as described in the same literature except the plates were incubated at 75 °C for 6 days.
2.7. Spontaneous Mutation Analysis
The rates of mutations that inactivate the pyrE gene were determined by previously described methods [26]. The overnight culture of the pyrE-proficient strains of DP-1 (OD600 = 0.67–0.69, 1.54–1.88 × 109 cells/mL) was diluted into 6 mL of fresh XT medium to yield a cell density of 5 × 103 cells/mL. The resulting cultures were incubated at 75 °C until they reached OD600= 0.1 with shaking. The same procedure was performed for strain HM-8 (OD600 = 0.61–0.66, 1.1–1.8 × 109 cells/mL). Each resulting culture was diluted 106–108-fold and spread on XTU plates, and was also spread on XTUF plates without dilution (in triplicate). The plates were incubated at 75 °C for 5 days. The mutation rate was calculated by the numbers of colonies that appeared on the plates.
3. Results
3.1. Deletion of PolB1-Binding Protein Genes
The MONSTER unmarked gene deletion method [23] was applied to the pbp1 and pbp2 genes of S. acidocaldarius. After transformation, 18 colonies/µg MONSTER-pbp1 were grown. No colony representing the pbp2 deletion strain could be isolated using either MONSTER-pbp2, which is constructed with an 84 bp deletion, or MONSTER-pbp2n, which is constructed with a 39 bp deletion. One colony showed blue color with X-gal solution was purified and analyzed its genotype by PCR using the outer primers and named strain HM-8 Int (Figure 1a). A total of 8.6 × 107 HM-8 Int cells were then applied for pop-out recombination using XTUF plate, and five white colonies were randomly selected. The genotypes of these colonies exhibited the expected deletion of approximately 0.2 kb in the pbp1 locus (Figure 1a). We also checked the deletion of the pbp1 gene using sequencing analysis and confirmed the expected 186 bp deletion in the pbp1 locus (data not shown). Thus, one correct pbp1 deletion strain was designated S. acidocaldarius strain HM-8 (Δpbp1). In addition, PCR analysis using inner primers yielded no product from HM-8 DNA (Figure 1b), indicating that the pbp1 gene was deleted from the original genomic locus and was not translocated.
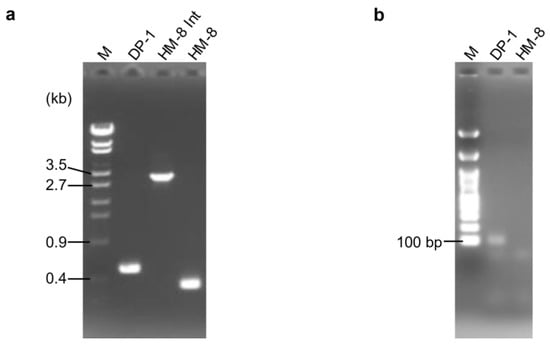
Figure 1.
PCR analysis of the pbp1 gene locus. (a) PCR analysis of the pbp1 locus of the S. acidocaldarius DP-1, intermediate (Int), and HM-8 strains using pbp1-out-F/R as primers. The expected sizes of the PCR bands were 0.5 kb (DP-1), 3 kb (HM-8 Int), and 0.4 kb (Δpbp1). A λ-EcoT14 ladder was loaded in lane M. (b) PCR analysis of the pbp1 locus of the S. acidocaldarius DP-1 and HM-8 strains using pbp1-in-F/R as primers. The expected sizes of the PCR bands were 87 bp (DP-1) and no band (Δpbp1). A 100-bp DNA ladder was loaded in lane M.
3.2. Growth Properties at Various Temperatures
The growth of deletion strain HM-8 (Δpbp1) was compared to that of the parent strain DP-1 over a wide temperature range (50–80 °C). At 80 °C, no growth of the Δpbp1 strain was observed, while the parent strain could grow (Figure 2). Between 50 °C and 75 °C, the growth of the Δpbp1 strain was nearly the same as that of the parent strain in the logarithmic growth phase (Figure S1).
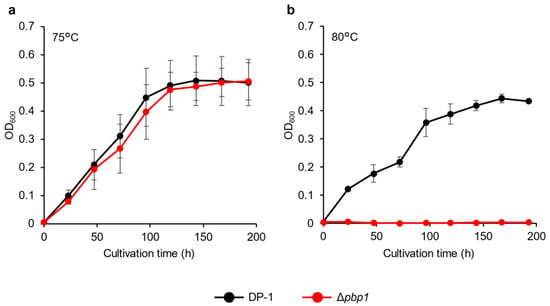
Figure 2.
Growth curves of the pbp1 deletion strain. Overnight cultures of the Δpbp1 (HM-8) and DP-1 strains were inoculated into xylose, tryptone, and uracil (XTU) liquid medium and cultivated at 75 °C (a) and 80 °C (b) without shaking. The error bars indicate the mean ± SD, calculated from triplicate experiments. Black line: The growth of DP-1; red line: The growth of Δpbp1 (HM-8).
3.3. Sensitivity to UV Irradiation
The growth of Δpbp1 after UV-B irradiation (zero, 400, 800, 1200, and 1600 J/m2) was characterized. The growth curves of Δpbp1 and the parent strain without irradiation were nearly the same (Figure 3). After UV irradiation at 400 J/m2, slight growth retardation of Δpbp1 was observed (Figure S2a). This retardation was clearer after UV irradiation at 800 J/m2 (Figure S2b). The difference became more striking after UV irradiation at 1200 (Figure 3) and 1600 J/m2 (Figure S2c). The results indicated that Δpbp1 exhibited significant sensitivity to helix-distorting lesions such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidine photoproducts (6-4PP) induced by UV irradiation [28,29,30].
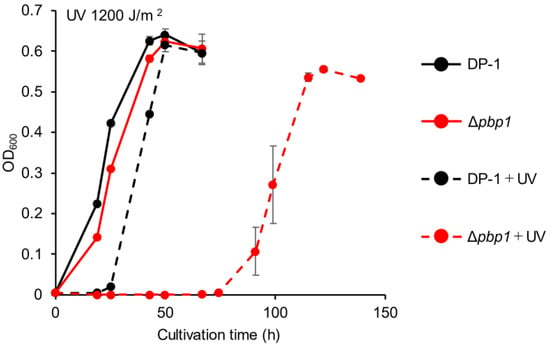
Figure 3.
Growth of the pbp1 deletion strain after UV-B irradiation. Overnight cultures of the Δpbp1 (HM-8) and DP-1 strains were irradiated with UV for 60 s (1200 J/m2) and cultivated at 75 °C with shaking. +UV represents a UV-treated sample. The error bars indicate the mean ± SD calculated from triplicate experiments. Black line: The growth of DP-1; red line: The growth of Δpbp1 (HM-8).
3.4. Sensitivity to Chemical Mutagens
The sensitivity of Δpbp1 to other helix-distorting lesions was also tested. Δpbp1 and the parent strain were incubated in growth medium with or without cisplatin (Wako, Chuo-Ku, Osaka, Japan) (70 and 100 μM). In the presence of cisplatin, the growth of Δpbp1 was the same as that of the parent strain (Figure 4a, Figure S3a). The growth of Δpbp1 was also tested in the presence or absence of 4-nitroquinoline N-oxide (4-NQNO) (TCI, Tokyo, Tokyo, Japan) (1 and 2 μM). In the presence of 1 μM 4-NQNO, the growth of Δpbp1 was retarded compared with that of the parent strain (Figure S3b). At 2 μM, the difference became more striking (Figure 4b). These results indicated that Δpbp1 exhibited significant sensitivity to bulky adducts by induced 4-NQNO, but did not show increased sensitivity to DNA intra strand and inter strand cross-links induced by cisplatin.
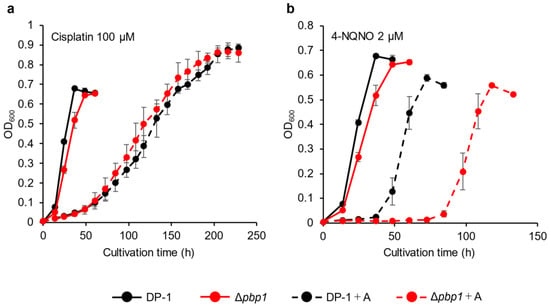
Figure 4.
Growth of the pbp1 deletion strain in the presence of DNA-damaging agents. Overnight cultures of the Δpbp1 (HM-8) and DP-1 strains were inoculated into liquid medium in the presence of DNA-damaging agents (cisplatin (100 μM (a)) and 4-nitroquinoline N-oxide (4-NQNO) (2 μM (b)) and cultivated at 75 °C with shaking. +A represents the growth with DNA-damaging agents. The error bars indicate the mean ± SD, calculated from triplicate experiments. Black line: The growth of DP-1; red line: The growth of Δpbp1 (HM-8).
To analyze the sensitivity of Δpbp1 to mitomycin C (MMC) (Wako, Chuo-Ku, Osaka, Japan), mock- and MMC-treated (zero, 180, 240, and 300 μM) aliquots of Δpbp1 and the parent strain were spotted on plates. No sensitivity of Δpbp1 to MMC (180 and 240 μM) was observed (Figure 5a). At 300 μM, the survival of Δpbp1 was slightly decreased in comparison with that of the parent strain (Figure 5a). The results suggested that Δpbp1 exhibited slight sensitivity to DNA inter strand crosslinks induced by MMC.
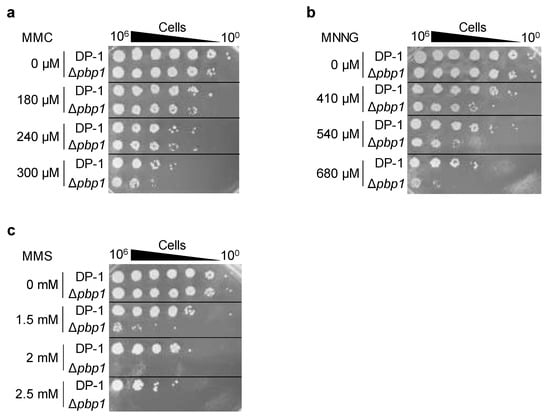
Figure 5.
Sensistivity to mitomycin C (MMC), methylnitronitrosoguanidine (MNNG), and methyl methanesulfonate (MMS) of the pbp1 deletion strain. DP-1 and Δpbp1 (HM-8) strains were treated with (a) MMC (0, 180, 240, and 300 μM), (b) MNNG (0, 410, 540, and 680 μM), and (c) MMS (0, 1.5, 2, and 2.5 mM), diluted (100–10−6), spotted onto XTU plates and cultivated at 75 °C.
To examine additional types of DNA damage, the cells of Δpbp1 and the parent strain were treated with methylnitronitrosoguanidine (MNNG) (SIGMA, Kawasaki, Kanagawa, Japan) and methyl methanesulfonate (MMS) (Wako, Chuo-Ku, Osaka, Japan) and were spotted on plates. The survival of Δpbp1 treated with MNNG (410 μM) was decreased compared to that of the parent strain, and this difference became more striking at 540 and 680 μM (Figure 5b). The survival of Δpbp1 after treatment with MMS (1.5 mM) was dramatically decreased in comparison with that of the parent strain, and this difference also became more striking at 2 and 2.5 mM (Figure 5c). These results indicated that Δpbp1 exhibited sensitivity to methylated base induced by MNNG or MMS. In particular, Δpbp1 showed greater sensitivity to 7-methylguanine and 3-methyladenine induced by MMS than O6-methylguanine induced by MNNG [31].
3.5. Sensitivity to Heat-Shock Treatment
The aliquots of Δpbp1 and the parent strain were heated at 90 °C for 0–4 min and spotted onto XTU plates. The survival of Δpbp1 was dramatically less than that of the parent strain after 2 min at 90 °C (Figure 6). This difference became more striking at longer heating times (3 or 4 min) (Figure 6). The results indicated that Δpbp1 was significantly sensitive to heat shock, which accelerates such reactions as follows (e.g., deamination, methylation, oxidation, and the formation of apurinic/apyrimidinic sites (AP sites)).
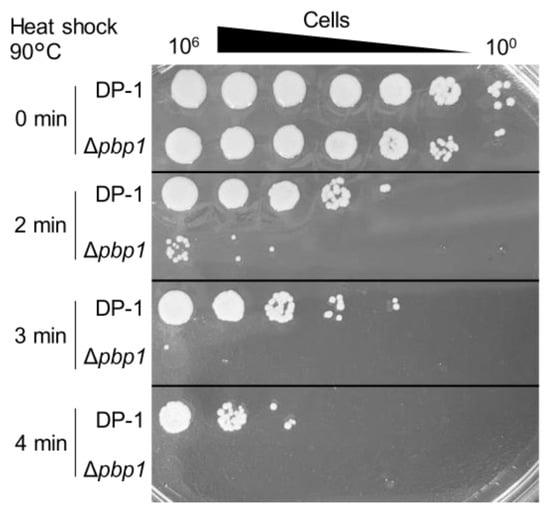
Figure 6.
Heat-shock sensitivity of the pbp1 deletion strain. After heat-shock at 90 °C for 0–4 min, diluted samples (10−6–100) of the DP-1 and Δpbp1 (HM-8) strains were spotted onto XTU plates and cultivated at 75 °C.
3.6. Sensitivity to DNA Replication Inhibitors
The growth of Δpbp1 in the presence of novobiocin (1.5 μM) was retarded compared with that of the parent strain (Figure S4a). The difference became more striking in the presence of novobiocin (3 (Figure 7a), 4.5 (Figure S4b), and 6 μM (Figure S4c)). In the presence of HU (25 μM), the growth of Δpbp1 was nearly the same as that of the parent strain (Figure S5a). In the presence of HU (50 μM), the growth of Δpbp1 was slightly delayed compared to the parent strain (Figure 7b). However, the growth of Δpbp1 was the same as that of the parent strain in the presence of HU (75 (Figure 7c) and 100 μM (Figure S5b)). These results indicated that Δpbp1 was highly sensitive to novobiocin. Novobiocin, a well-known topoisomerase inhibitors in bacteria and/or eukaryotes, was reported to slow down or arrest chromosome replication at elongation stage in S. acidocaldarius [32]. On the other hand, Δpbp1 did not exhibit sensitivity to HU in this study in contrast to the chromosome replication that was perturbed in S. solfataricus by an unknown mechanism [33].
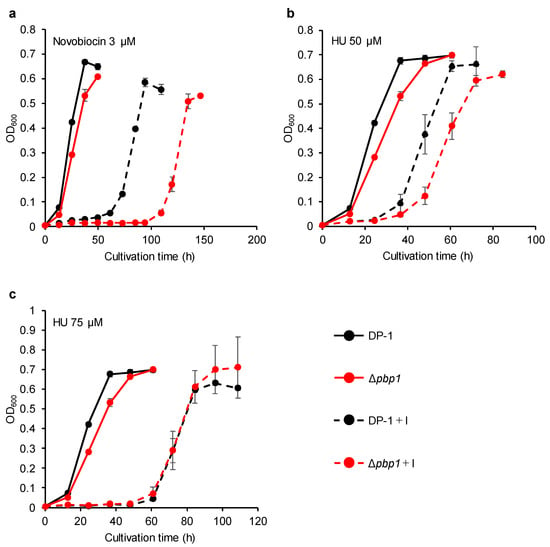
Figure 7.
Growth of the pbp1 deletion strain in the presence of DNA replication inhibitors. Overnight cultures of the Δpbp1 (HM-8) and DP-1 strains were inoculated into liquid medium in the presence of a DNA replication inhibitor (novobiocin (3 μM (a)) and HU (50 (b) and 75 μM (c))) and cultivated at 75 °C with shaking. +I represents the growth with a DNA replication inhibitor. The error bars indicate the mean ± SD, calculated from triplicate experiments. Black line: The growth of DP-1; red line: The growth of Δpbp1 (HM-8).
3.7. Estimation of Mutation Rates
We investigated the mutation frequency of Δpbp1. Mutation assays revealed that the mutation rate of Δpbp1 was 10-fold higher than that of the parent strain (4.3 (±0.2) ×10−5 for the parent strain vs. 3.3 (±0.8) ×10−4 for Δpbp1). The results indicated that PBP1 is important for mutation avoidance.
4. Discussion
To examine whether PBP1 and PBP2 are essential for the activity of apo-PolB1, we attempted to delete the pbp1 and pbp2 genes independently in S. acidocaldarius. As a result, a pbp1 deletion strain was constructed; however, no pbp2 deletion strain was isolated. These results demonstrated that PBP2 is essential for DNA replication by apo-PolB1. In addition, Δpbp1 exhibited sensitivity to numerous types of DNA damage, suggesting that PBP1 is important in DNA repair or the tolerance of DNA damage by apo-PolB1.
PolB1 has been found in all members of the TACK (Thaumarchaota, Aigarchaota, Crenarchaeota, and Korarchaeota) superphylum of Archaea [10,11]. PBP1 and PBP2 are present in the order Sulfolobales, Acidilobales, and Desulfurococcales, which belong to Crenarchaea [11,20]. On the other hand, Thaumarchaea, Aigarchaea, Korarchaea, and Crenarchaea of the order Thermoproteales do not possess homologs of PBP1 or PBP2 [11,20]. Thaumarchaea, Aigarchaea, and Korarchaea also possess D-family polymerase, which is a replicative polymerase, in addition to PolB1, while Crenarchaea of the order Thermoproteales lack a D-family polymerase [11,20]. Almost all Thermoproteales have acidic extensions in the N-terminal regions of PolB1, which may serve as alternatives playing the role of PBP1 [20]. Similarly, the alternatives playing the role of PBP2 may be present in Thermoproteales since PBP2 is essential for the activity of PolB1 in S. acidocaldarius.
The development and application of PCR technology using thermophilic bacterial and archaeal DNAPs has been considered. B-family polymerases of archaea such as Pyrococcus furiosus, Thermococcus kodakarensis, and Thermococcus litralis are often used as PCR enzymes [34,35,36]. B-family polymerases of Sulfolobales have not been practically used for PCR, but attempts have been made to apply them for PCR. The suitability of PolB3, but not PolB1 for PCR, has been verified in Crenarchaea [37,38,39,40]. This may be attributed to the absence of PBP2, which is essential for replication by apo-PolB1. In addition, holoenzyme PolB1 in S. solfataricus is capable of performing PCR [20].
In this study, the growth of the Δpbp1 strain was nearly the same as that of the parent strain at 75 °C (optimal growth condition), although PBP1 is important for lagging strand DNA synthesis [20]. In addition, Δpbp1 exhibited sensitivity to various types of DNA damage, suggesting that PBP1 is involved in DNA repair or damage tolerance rather than lagging strand synthesis by apo-PolB1. A previous in vitro study reported that two chromatin proteins, Sso7d (Sul7d) and Cren7, inhibited the robust strand displacement by apo-PolB1 in S. solfataricus [41]. Sul7d is highly conserved in Sulfolobus [42], whereas Cren7 (an essential gene in Sulfolobus islandicus [43]) is widely conserved in Crenarchaea, except for Thermophilum pendens [44]. Taken together, Cren7 and Sul7d, but not PBP1, are mainly responsible for inhibiting excessive strand displacement by apo-PolB1 during Okazaki fragment maturation [41]. DNA repair by apo-PolB1 is possibly enabled by inhibiting excessive displacement of apo-PolB1 during gap filling. Bacteria have PolI, which is an A-family polymerase and is involved in the maturation of the Okazaki fragments at the lagging strand [5]. PolI has 5′ to 3′ exonuclease activity to remove the ribonucleotide portion of newly synthesized Okazaki fragments and DNA polymerase activity to fill in the resulting gap [45]. In addition, PolI fills in DNA gaps that result from the removal of a variety of DNA lesions (e.g., the UV-induced thymidine dimer, the oxidative lesion 8-oxoguanine, and the alkylation lesion 4-methyladenine) during repair [45]. Holoenzyme PolB1 in archaea seems to play the roles of both PolI, which removes RNA primers and fills the gap in DNA repair, and PolIII, which replicates leading and lagging strands in bacteria.
In this study, no growth of Δpbp1 was observed at 80°C. Genetic evidence indicates that PBP1 is important for the thermostability of apo-PolB1, consistent with a previous in vitro study showing that holoenzyme PolB1 in the presence of PBP1 and PBP2 causes a large increase in the thermostability of the enzyme compared to apo-PolB1 [20]. Our results showed that Δpbp1 exhibited high sensitivity to various types of damage, suggesting that holoenzyme PolB1 contributes to DNA repair or to the tolerance of broad types of DNA damage. In particular, Δpbp1 is substantially sensitive to UV irradiation, MMS, 4-NQNO, heat shock, and novobiocin. S. acidocaldarius has three accessory DNAPs, namely, PolB2, PolB3, and Dbh. These deletion strains, including double and triple mutants, did not exhibit sensitivity to MMS compared with the parent strain [15]. In addition, these deletion strains were not sensitive to novobiocin at 75 °C [15]. This indicates that holoenzyme PolB1 rather than three accessory DNAPs mainly contributes to the repair or tolerance of damage induced by MMS and novobiocin. A previous in vivo study indicated that the ΔpolB2 ΔpolB3 combination was sensitive to UV, but the effect was limited in magnitude [15]. This study showed that Δpbp1 exhibited significant sensitivity to UV irradiation, suggesting that holoenzyme PolB1 is mainly involved in the repair or tolerance of UV damage rather than PolB2, PolB3, and Dbh. The DNA damage induced by heat shock (e.g., deamination, methylation, oxidation, and the formation of AP sites) and methylated base induced by MNNG and MMS are thought to be repaired by base excision repair (BER) or alternative excision repair (AER) [2,3,46]. On the other hand, helix-distorting DNA lesions such as CPDs, 6-4PP, and bulky adducts induced by UV irradiation and 4-NQNO are thought to be repaired by the nucleotide excision repair (NER) [2,3,47]. It is suggested that PBP1 is involved in gap filling by holoenzyme PolB1 in these DNA repair pathways. However, thermophilic archaea are known to lack some NER proteins, so the mechanism by which helix-distorting DNA damage is repaired is interesting, but unknown in archaea [3,47,48,49]. On the other hand, Δpbp1 was not sensitive (or was slightly sensitive) to MMC and cisplatin, which induce inter strand DNA crosslinks. In addition, Δpbp1 did not exhibit sensitivity to HU, which is an inhibitor of DNA synthesis. Although the mechanism of repair of inter strand cross-linking is not well understood in archaea, PBP1 may not be involved directly in the repair of inter strand cross-linking.
Interaction with PBP1 reduced 3′ to 5′ exonuclease activity compared to that of apo-PolB1 [20]. It was speculated that 3′ to 5′ exonucleolytic proofreading was promoted in the absence of PBP1 in vivo. However, the mutation rate of Δpbp1 was significantly increased compared to that of the parent strain. These results suggested that inhibition of the 3′ to 5′ exonuclease activity by PBP1 had no direct influence on accurate replication, but indicated that the effects of proofreading by holoenzyme PolB1 may be complicated. A moderate 3′ to 5′ exonuclease activity is probably necessary for DNA integrity.
5. Conclusions
To examine whether PBP1 and PBP2 are essential for the activity of apo-PolB1 in S. acidocaldarius, we attempted to delete the pbp1 and pbp2 genes independently. It was possible to construct a Δpbp1 strain, but not a Δpbp2 strain. In addition, Δpbp1 exhibited high sensitivity to various types of damage and an increased mutation rate. In particular, Δpbp1 exhibited greater sensitivity to UV irradiation, MMS, and novobiocin than the deletion strains of polB2, polB3, and dbh, including double and triple mutants [15]. These results suggested that holoenzyme PolB1 contributes to both replication and repair. PBP1 is involved in the repair or tolerance of various types of DNA damage, although it is not essential for the activity of apo-PolB1. On the other hand, PBP2 is essential for replication by apo-PolB1. Thus, holoenzyme PolB1 of S. acidocaldarius is versatile. These results provide new genetic evidence of the biological function of holoenzyme PolB1.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/9/2/439/s1, Figure S1. Growth curves of the pbp1 deletion strain. Figure S2. Growth of the pbp1 deletion strain after UV-B irradiation. Figure S3. Growth of the pbp1 deletion strain in the presence of DNA-damaging agents. Figure S4. Growth of the pbp1 deletion strain in the presence of novobiocin. Figure S5. Growth of the pbp1 deletion strain in the presence of HU.
Author Contributions
Conceptualization and methodology, H.M. and N.K.; formal analysis and investigation, H.M.; resources and data curation, H.D.S. and N.K.; writing—original draft preparation, H.M.; writing—review and editing, H.D.S. and N.K.; supervision, project administration, and funding acquisition, N.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Special Grant of the Faculty of Science and Engineering, Soka University (to H.D.S. and NK).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, and further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Johansson, E.; Dixon, N. Replicative DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012799. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. DNA repair. In Archaea: Evolution, Physiology and Molecular Biology; Garrett, R.A., Klenk, H.P., Eds.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2007; pp. 171–183. [Google Scholar]
- White, M.F.; Allers, T. DNA repair in the archaea–an emerging picture. FEMS Microbiol. Rev. 2018, 42, 514–526. [Google Scholar] [CrossRef]
- McHenry, C.S. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011, 15, 587–594. [Google Scholar] [CrossRef]
- Raia, P.; Delarue, M.; Sauguet, L. An updated structural classification of replicative DNA polymerases. Biochem. Soc. Trans. 2019, 47, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Burgers, P.M. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008, 18, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, F.; Long, F.; Cann, I.; Whitman, W.B. Diversity of the DNA replication system in the archaea domain. Archaea 2014, 2014, 675946. [Google Scholar] [CrossRef]
- Doublie, S.; Zahn, K.E. Structural insights into eukaryotic DNA replication. Front. Microbiol. 2014, 5, 444. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Makarova, K.S.; Krupovic, M.; Koonin, E.V. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front. Microbiol. 2014, 5, 354. [Google Scholar] [CrossRef]
- Cooper, C.D.O. Archaeal DNA polymerases: New frontiers in DNA replication and repair. Emerg. Top. Life Sci. 2018, 2, 503–516. [Google Scholar] [CrossRef]
- Cubonová, L.; Richardson, T.; Burkhart, B.W.; Kelman, Z.; Connolly, B.A.; Reeve, J.N.; Santangelo, T.J. Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J. Bacteriol. 2013, 195, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, F.; Mrázek, J.; Whitman, W.B. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. USA 2013, 110, 4726–4731. [Google Scholar] [CrossRef] [PubMed]
- Kushida, T.; Narumi, I.; Ishino, S.; Ishino, Y.; Fujiwara, S.; Imanaka, T.; Higashibata, H. Pol B, a family B DNA polymerase, in Thermococcus kodakarensis is important for DNA repair, but not DNA replication. Microbes Environ. 2019, 34, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, H.; Jain, R.; Suzuki, S.; Grogan, D.W.; Kurosawa, N. PolB1 is sufficient for DNA replication and repair under normal growth conditions in the extremely thermophilic crenarchaeon Sulfolobus acidocaldarius. Front. Microbiol. 2020, 11, 613375. [Google Scholar] [CrossRef]
- Feng, X.; Liu, X.; Xu, R.; Zhao, R.; Feng, W.; Liao, J.; Han, W.; She, Q. A unique B-Family DNA polymerase facilitating error-prone DNA damage tolerance in Crenarchaeota. Front. Microbiol. 2020, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Cann, I.K.O.; Komori, K.; Toh, H.; Kanai, S.; Ishino, Y. A heterodimeric DNA polymerase: Evidence that members of Euryarchaeota possess a distinct DNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95. [Google Scholar] [CrossRef] [PubMed]
- Sauguet, L.; Raia, P.; Henneke, G.; Delarue, M. Shared active site architecture between archaeal PolD and multi-subunit RNA polymerases revealed by X-ray crystallography. Nat. Commun. 2016, 7, 12227–12238. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Grummt, F.; Burger, K.J. Purification and characterization of DNA polymerase from the archaebacterium Sulfolobus acidocaldarius. Nucleic Acids Res. 1985, 13, 5269–5282. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Beattie, T.R.; Rojas, A.L.; Schermerhorn, K.; Gristwood, T.; Trinidad, J.C.; Albers, S.V.; Roversi, P.; Gardner, A.F.; Abrescia, N.G.A.; et al. Identification and characterization of a heterotrimeric archaeal DNA polymerase holoenzyme. Nat. Commun. 2017, 8, 15075. [Google Scholar] [CrossRef]
- Sakai, H.D.; Kurosawa, N. Saccharolobus caldissimus gen. nov., sp. nov., a facultatively anaerobic iron-reducing hyperthermophilic archaeon isolated from an acidic terrestrial hot spring, and reclassification of Sulfolobus solfataricus as Saccharolobus solfataricus comb. nov. and Sulfolobus shibatae as Saccharolobus shibatae comb. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 1271–1278. [Google Scholar] [CrossRef]
- Cranford, M.T.; Kaszubowski, J.D.; Trakselis, M.A. A hand-off of DNA between archaeal polymerases allows high-fidelity replication to resume at a discrete intermediate three bases past 8-oxoguanine. Nucleic Acids Res. 2020, 48, 10986–10997. [Google Scholar] [CrossRef]
- Suzuki, S.; Kurosawa, N. Development of the multiple gene knockout system with one-step PCR in thermoacidophilic crenarchaeon Sulfolobus acidocaldarius. Archaea 2017, 2017, 7459310. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kurosawa, N. Disruption of the gene encoding restriction endonuclease SuaI and development of a host-vector system for the thermoacidophilic archaeon Sulfolobus acidocaldarius. Extremophiles 2016, 20, 139–148. [Google Scholar] [CrossRef]
- Grogan, D.W. Isolation of Sulfolobus acidocaldarius mutants. In Archaea: A Laboratory Manual-Thermophiles; Robb, F.T., Place, A.R., Sowers, K.R., Schreier, H.J., DasSarma, S., Fleishmann, E.M., Eds.; CSH Press: Cold Spring Harbor, NY, USA, 1995; pp. 125–132. [Google Scholar]
- Suzuki, S.; Kurosawa, N. Endonucleases responsible for DNA repair of helix-distorting DNA lesions in the thermophilic crenarchaeon Sulfolobus acidocaldarius in vivo. Extremophiles 2019, 23, 613–624. [Google Scholar] [CrossRef]
- Reilly, M.S.; Grogan, D.W. Characterization of intragenic recombination in a hyperthermophilic archaeon via conjugational DNA exchange. J. Bacteriol. 2001, 183, 2943–2946. [Google Scholar] [CrossRef]
- Courcelle, J.; Crowley, D.J.; Hanawalt, P.C. Recovery of DNA replication in UV-irradiated Escherichia coli requires both excision repair and RecF protein function. J. Bacteriol. 1999, 181, 916–922. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Dorazi, R.; Götz, D.; Munro, S.; Bernander, R.; White, M.F. Equal rates of repair of DNA photoproducts in transcribed and non-transcribed strands in Sulfolobus solfataricus. Mol. Microbiol. 2007, 63, 521–529. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Hjort, K.; Bernander, R. Cell cycle regulation in the hyperthermophilic crenarchaeon Sulfolobus acidocaldarius. Mol. Microbiol. 2001, 40, 225–234. [Google Scholar] [CrossRef]
- Liew, L.P.; Lim, Z.Y.; Cohen, M.; Kong, Z.; Marjavaara, L.; Chabes, A.; Bell, S.D. Hydroxyurea-mediated cytotoxicity without inhibition of ribonucleotide reductase. Cell Rep. 2016, 17, 1657–1670. [Google Scholar] [CrossRef]
- Cariello, N.F.; Swenberg, J.A.; Skopek, T.R. Fidelity of Thermococcus litoralis DNA polymerase (VentTM) in PCR determined by denaturing gradient gel electrophoresis. Nucleic Acids Res. 1991, 19, 4193–4198. [Google Scholar] [CrossRef]
- Lundberg, K.S.; Shoemaker, D.D.; Adams, M.W.; Short, J.M.; Sorge, J.A.; Mathur, E.J. High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 1991, 108, 1–6. [Google Scholar] [CrossRef]
- Takagi, M.; Nishioka, M.; Kakihara, H.; Kitabayashi, M.; Inoue, H.; Kawakami, B.; Oka, M.; Imanaka, T. Characterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl. Environ. Microbiol. 1997, 63, 4504–4510. [Google Scholar] [CrossRef]
- Kähler, M.; Antranikian, G. Cloning and characterization of a family B polymerase from the hyperthermophilic crenarchaeon Pyrobaculum islandicum. J. Bacteriol. 2000, 182, 655–663. [Google Scholar] [CrossRef]
- Seo, K.J.; Cho, S.S.; Ppyun, H.W.; Youn, M.H.; Kim, S.H.; Seo, B.S.; Kwon, S.T. Characterization of a family B DNA polymerase from the hyperthermophilic crenarchaeon Ignicoccus hospitalis KIN4/I and its application to PCR. Appl. Biochem. Biotechnol. 2014, 173, 1108–1120. [Google Scholar] [CrossRef]
- Daimon, K.; Ishino, S.; Imai, N.; Nagumo, S.; Yamagami, T.; Matsukawa, H.; Ishino, Y. Two family B DNA polymerases from Aeropyrum pernix, based on revised translational frames. Front. Mol. Biosci. 2018, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T. Functional and phylogenetic analysis of DNA polymerase and cofactor PCNA in Crenarchaeota. Ph.D. Thesis, Soka University, Tokyo, Japan, 2001. [Google Scholar]
- Sun, F.; Huang, L. Sulfolobus chromatin proteins modulate strand displacement by DNA polymerase B1. Nucleic Acids Res. 2013, 41, 8182–8195. [Google Scholar] [CrossRef]
- Choli, T.; Henning, P.; Wittmann-Liebold, B.; Reinhardt, R. Isolation, characterization and microsequence analysis of a small basic methylated DNA-binding protein from the Archaebacterium, Sulfolobus solfataricus. Biochim. Biophys. Acta 1988, 950, 193–203. [Google Scholar] [CrossRef]
- Zhang, C.; Phillips, A.P.R.; Wipfler, R.L.; Olsen, G.J.; Whitaker, R.J. The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat. Commun. 2018, 9, 4908. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Feng, Y.; Zhang, Z.; Yao, H.; Luo, Y.; Wang, J.; Huang, L. Biochemical and structural characterization of Cren7, a novel chromatin protein conserved among Crenarchaea. Nucleic Acids Res. 2007, 36, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.H.; Suzuki, M.; Adman, E.; Shinkai, A.; Loeb, L.A. Prokaryotic DNA polymerase I: Evolution, structure, and “base flipping” mechanism for nucleotide selection. J. Mol. Biol. 2001, 308, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Yasui, A. Alternative Excision Repair Pathways. Cold Spring Harb. Perspect. Biol. 2013, 5, a012617. [Google Scholar] [CrossRef]
- Grogan, D.W. Understanding DNA repair in hyperthermophilic archaea: Persistent gaps and other reactions to focus on the fork. Archaea 2015, 2015, 942605. [Google Scholar] [CrossRef]
- Fujikane, R.; Ishino, S.; Ishino, Y.; Forterre, P. Genetic analysis of DNA repair in the hyperthermophilic archaeon, Thermococcus kodakarensis. Genes Genet. Syst. 2010, 85, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, B.; Li, S.; Ao, X.; Dalgaard, K.; Gökce, S.; Liang, Y.; She, Q. Genetic manipulation in Sulfolobus islandicus and functional analysis of DNA repair genes. Biochem. Soc. Trans. 2013, 41, 405–410. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).