
Applications of Blocker Nucleic Acids and Non-Metazoan PCR Improves the Discovery of the Eukaryotic Microbiome in Ticks

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ticks and DNA Extraction
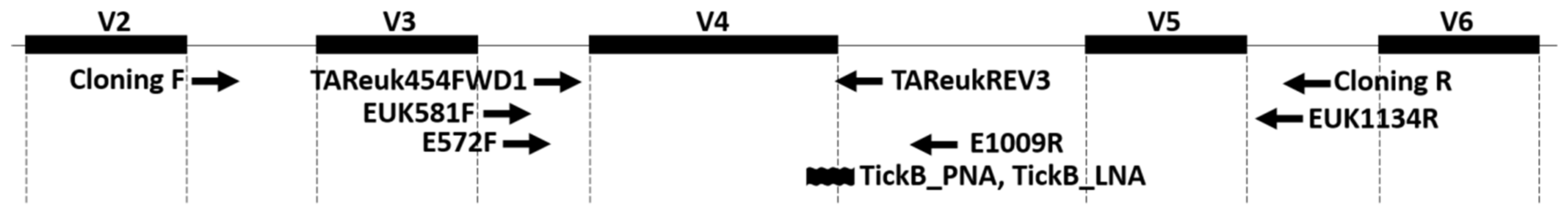
2.2. Design of Blocker Nucleic Acids
2.3. Plasmid Preparation
2.4. PCR Amplification with the Addition of TickB_PNA and TickB_LNA
2.5. UNonMet-PCR Amplification
2.6. Illumina Library Preparation and MiSeq Run
2.7. Data Processing and Analysis
3. Results
3.1. Designing of TickB_PNA and TickB_LNA
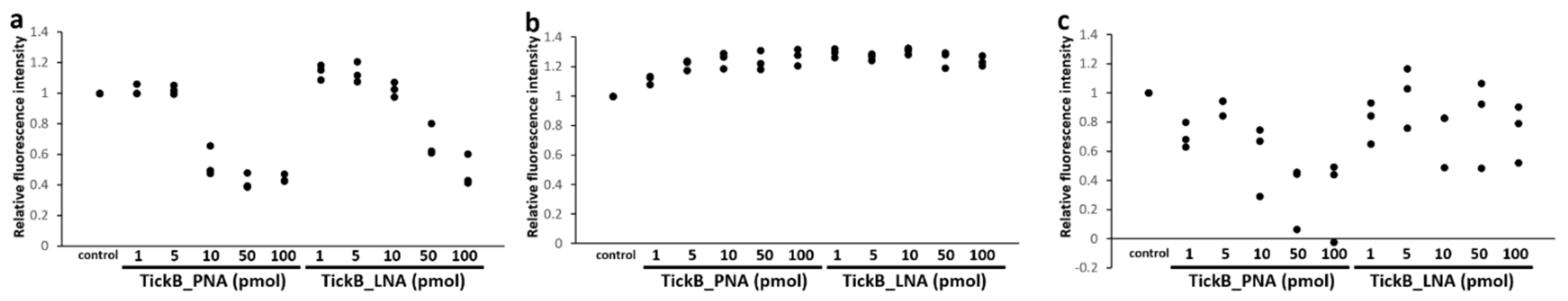
3.2. Blocking Efficacy of TickB_PNA and TickB_LNA
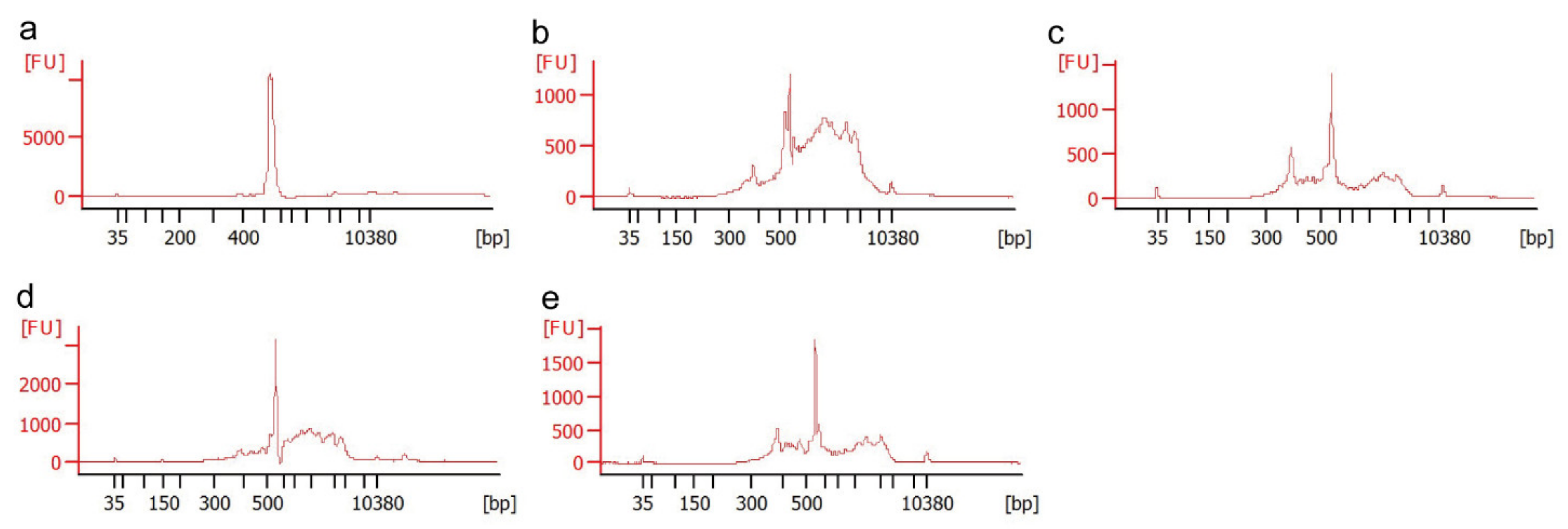
3.3. Comparison of Purification Methods by a Fragment Length Analysis
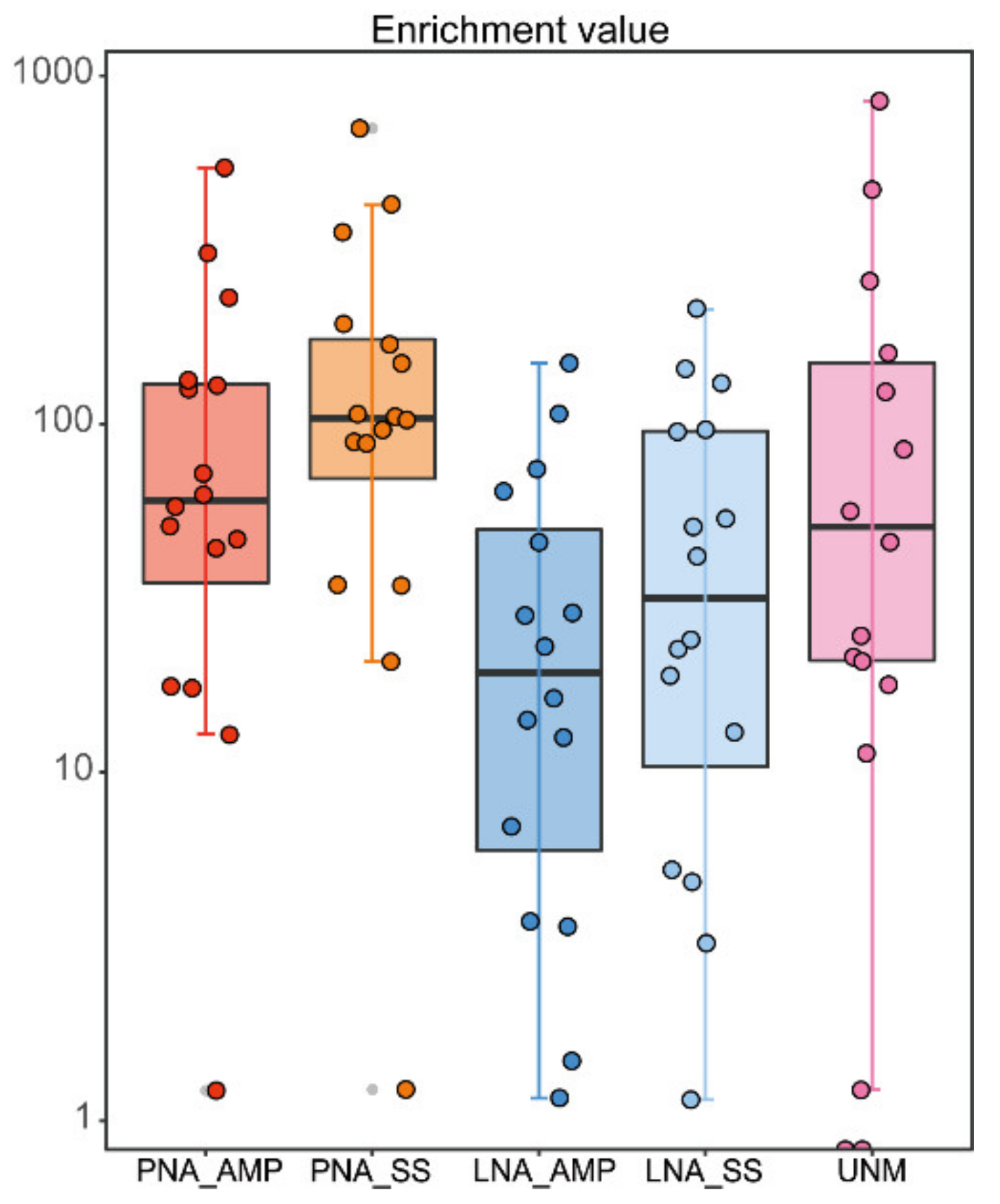
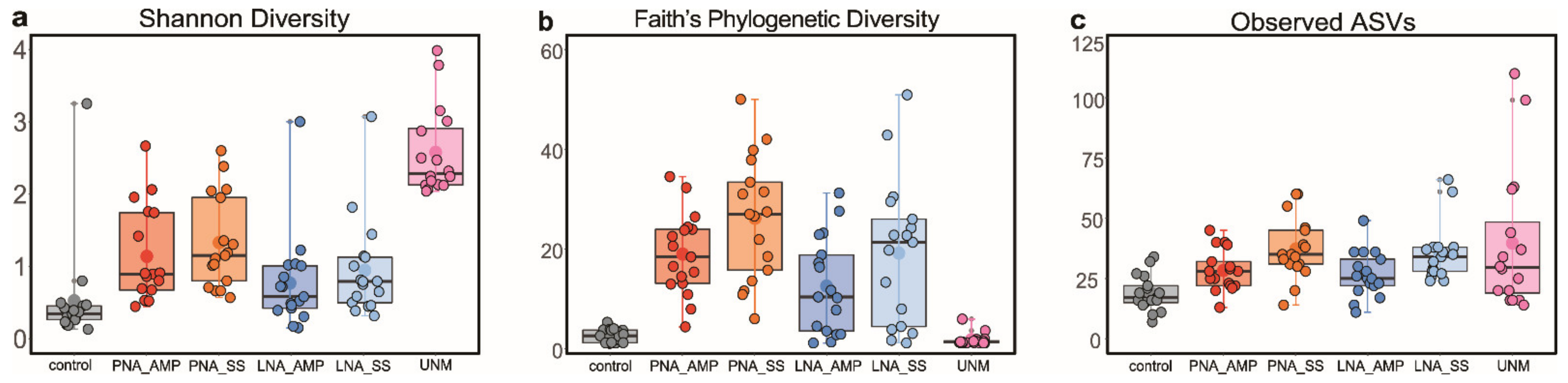
3.4. Comparison of Blocking Efficacy of TickB_PNA, TickB_LNA, and UNonMet-PCR
3.5. PCR with Blockers or UNonMet-PCR
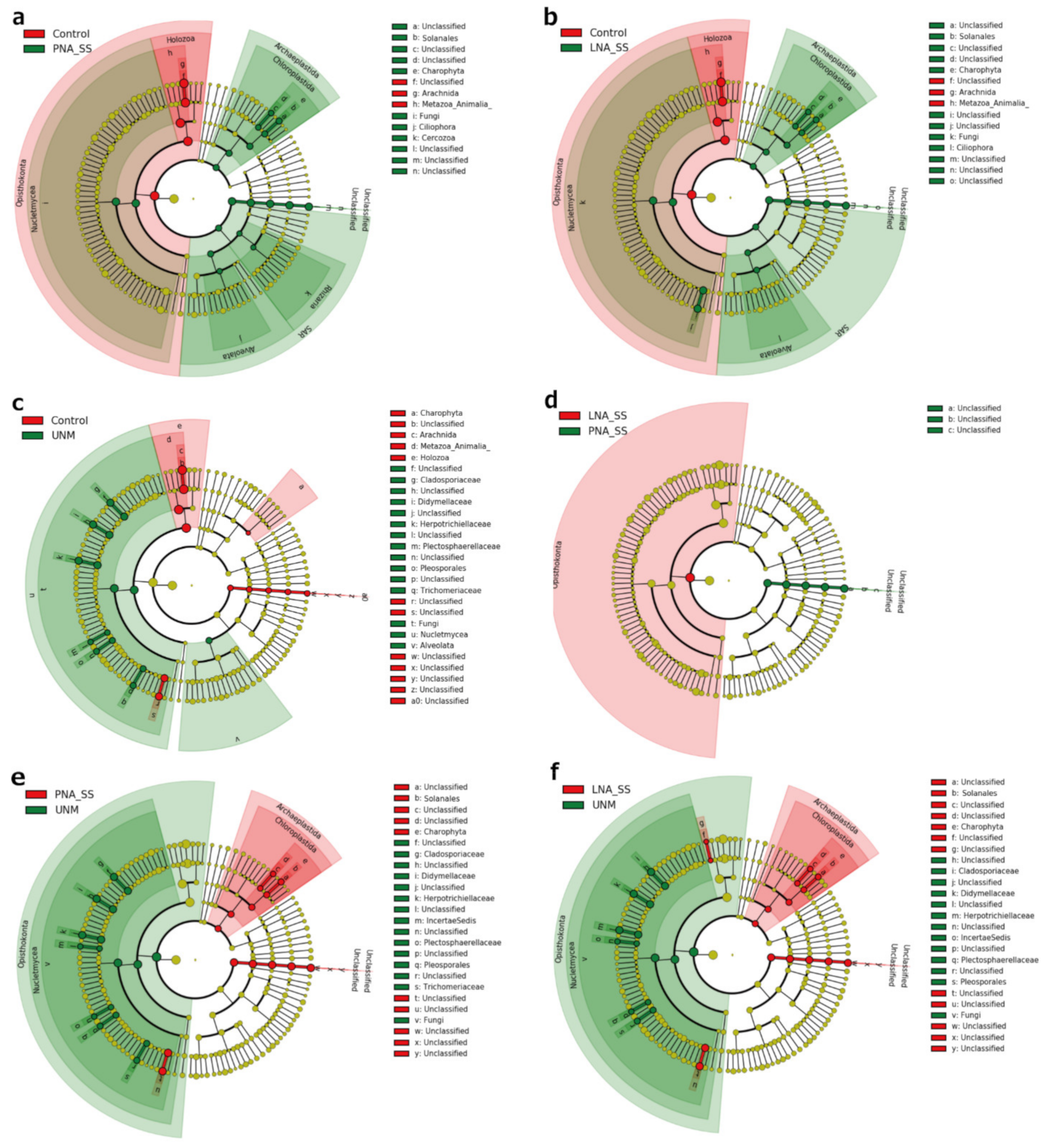
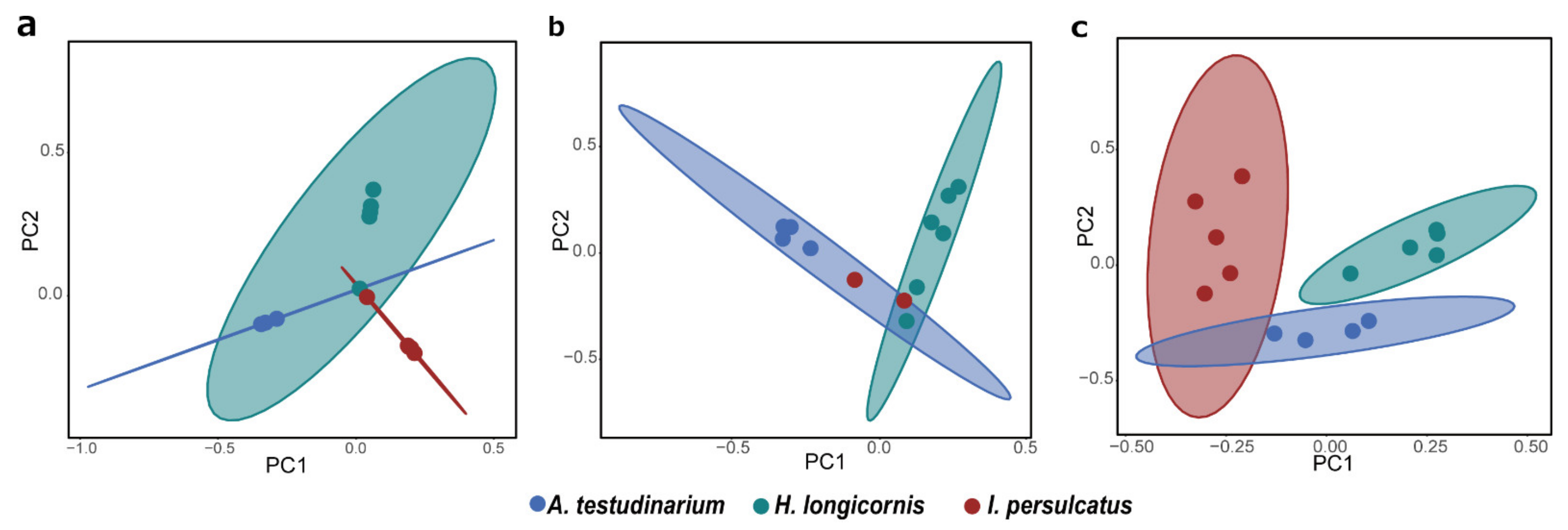
3.6. Comparison of Eukaryotic Microbiomes among Three Tick Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gottlieb, Y.; Lalzar, I.; Klasson, L. Distinctive Genome Reduction Rates Revealed by Genomic Analyses of Two Coxiella- Like Endosymbionts in Ticks. Genome Biol. Evol. 2015, 7, 1779–1796. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.J.; Torkelson, J.L.; Bodnar, J.; Mortazavi, B.; Laurent, T.; Deason, J.; Thephavongsa, K.; Zhong, J. The Rickettsia Endosymbiont of Ixodes Pacificus Contains All the Genes of De Novo Folate Biosynthesis. PLoS ONE 2015, 10, e0144552. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; DePonte, K.; Fish, D.; Fikrig, E. Gut Microbiota of the Tick Vector Ixodes Scapularis Modulate Colonization of the Lyme Disease Spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Steiner, F.E.; Pinger, R.R.; Vann, C.N.; Grindle, N.; Civitello, D.; Clay, K.; Fuqua, C. Infection and Co-Infection Rates of Anaplasma Phagocytophilum Variants, Babesia Spp., Borrelia Burgdorferi, and the Rickettsial Endosymbiont in Ixodes Scapularis (Acari: Ixodidae) from Sites in Indiana, Maine, Pennsylvania, and Wisconsin. J. Med. Entomol. 2008, 45, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Moriya, S.; Ohkuma, M.; Kudo, T. Molecular Cloning and Characterization of a Cellulase Gene from a Symbiotic Protist of the Lower Termite, Coptotermes Formosanus. Gene 2005, 349, 67–75. [Google Scholar] [CrossRef]
- Ohkuma, M. Symbioses of Flagellates and Prokaryotes in the Gut of Lower Termites. Trends Microbiol. 2008, 16, 345–352. [Google Scholar] [CrossRef]
- Watanabe, H.; Takase, A.; Tokuda, G.; Yamada, A.; Lo, N. Symbiotic “Archaezoa” of the Primitive Termite Mastotermes Darwiniensis Still Play a Role in Cellulase Production. Eukaryot. Cell 2006, 5, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, S.; Ohkuma, M.; Yamada, A.; Hongoh, Y.; Kudo, T. Phylogenetic Position and In Situ Identification of Ectosymbiotic Spirochetes on Protists in the Termite Gut. AEM 2003, 69, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Stingl, U.; Radek, R.; Yang, H.; Brune, A. “Endomicrobia”: Cytoplasmic Symbionts of Termite Gut Protozoa Form a Separate Phylum of Prokaryotes. AEM 2005, 71, 1473–1479. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, Y.; Moriyama, M.; Łukasik, P.; Vanderpool, D.; Tanahashi, M.; Meng, X.-Y.; McCutcheon, J.P.; Fukatsu, T. Recurrent Symbiont Recruitment from Fungal Parasites in Cicadas. Proc. Natl. Acad. Sci. USA 2018, 115, E5970–E5979. [Google Scholar] [CrossRef] [Green Version]
- Austen, J.M.; Ryan, U.M.; Friend, J.A.; Ditcham, W.G.F.; Reid, S.A. Vector of Trypanosoma Copemani Identified as Ixodes Sp. Parasitology 2011, 138, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Thekisoe, O.M.M.; Honda, T.; Fujita, H.; Battsetseg, B.; Hatta, T.; Fujisaki, K.; Sugimoto, C.; Inoue, N. A Trypanosome Species Isolated from Naturally Infected Haemaphysalis Hystricis Ticks in Kagoshima Prefecture, Japan. Parasitology 2007, 134, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Greengarten, P.J.; Tuininga, A.R.; Morath, S.U.; Falco, R.C.; Norelus, H.; Daniels, T.J. Occurrence of Soil- and Tick-Borne Fungi and Related Virulence Tests for Pathogenicity to Ixodes Scapularis (Acari: Ixodidae). J. Med. Entomol. 2011, 48, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Tuininga, A.R.; Miller, J.L.; Morath, S.U.; Daniels, T.J.; Falco, R.C.; Marchese, M.; Sahabi, S.; Rosa, D.; Stafford, K.C. Isolation of Entomopathogenic Fungi From Soils and Ixodes Scapularis (Acari: Ixodidae) Ticks: Prevalence and Methods. J. Med. Entomol. 2009, 46, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otranto, D.; Brianti, E.; Latrofa, M.S.; Annoscia, G.; Weigl, S.; Lia, R.P.; Gaglio, G.; Napoli, E.; Giannetto, S.; Papadopoulos, E.; et al. On a Cercopithifilaria Sp. Transmitted by Rhipicephalus Sanguineus: A Neglected, but Widespread Filarioid of Dogs. Parasites Vectors 2012, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Nakao, R.; Ohnuma, A.; Kawamori, F.; Sugimoto, C. Microbial Population Analysis of the Salivary Glands of Ticks; A Possible Strategy for the Surveillance of Bacterial Pathogens. PLoS ONE 2014, 9, e103961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swei, A.; Kwan, J.Y. Tick Microbiome and Pathogen Acquisition Altered by Host Blood Meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef]
- Thapa, S.; Zhang, Y.; Allen, M.S. Effects of Temperature on Bacterial Microbiome Composition in Ixodes Scapularis Ticks. Microbiologyopen 2019, 8, e00719. [Google Scholar] [CrossRef] [Green Version]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent Insights into the Tick Microbiome Gained through Next-Generation Sequencing. Parasites Vectors 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, M.-C.; Arévalo, A.; Stiemsma, L.; Dimitriu, P.; Chico, M.E.; Loor, S.; Vaca, M.; Boutin, R.C.T.; Morien, E.; Jin, M.; et al. Associations between Infant Fungal and Bacterial Dysbiosis and Childhood Atopic Wheeze in a Nonindustrialized Setting. J. Allergy Clin. Immunol. 2018, 142, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Belda, E.; Coulibaly, B.; Fofana, A.; Beavogui, A.H.; Traore, S.F.; Gohl, D.M.; Vernick, K.D.; Riehle, M.M. Preferential Suppression of Anopheles Gambiae Host Sequences Allows Detection of the Mosquito Eukaryotic Microbiome. Sci. Rep. 2017, 7, 3241. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical Innovations for High-Throughput Amplicon Sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Taerum, S.J.; Steven, B.; Gage, D.J.; Triplett, L.R. Validation of a PNA Clamping Method for Reducing Host DNA Amplification and Increasing Eukaryotic Diversity in Rhizosphere Microbiome Studies. Phytobiomes J. 2020, PBIOMES-05-20-0040-TA. [Google Scholar] [CrossRef]
- Reigel, A.M.; Owens, S.M.; Hellberg, M.E. Reducing Host DNA Contamination in 16S rRNA Gene Surveys of Anthozoan Microbiomes Using PNA Clamps. Coral Reefs 2020, 39, 1817–1827. [Google Scholar] [CrossRef]
- Briones, C.; Moreno, M. Applications of Peptide Nucleic Acids (PNAs) and Locked Nucleic Acids (LNAs) in Biosensor Development. Anal. Bioanal. Chem. 2012, 402, 3071–3089. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, M.; Sakai, M. Application of Locked Nucleic Acid (LNA) Oligonucleotide–PCR Clamping Technique to Selectively PCR Amplify the SSU RRNA Genes of Bacteria in Investigating the Plant-Associated Community Structures. Microb. Environ. 2014, 29, 286–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-K.; Klaus, D.L.; Retes, P.; Castro, P.; Castro, M. Bridged Nucleic Acids (BNAs) as Molecular Tools. J. Biochem. Mol. Biol. Res. 2015, 1, 67–71. [Google Scholar] [CrossRef]
- Carnegie, R.; Meyer, G.; Blackbourn, J.; Cochennec-Laureau, N.; Berthe, F.; Bower, S. Molecular Detection of the Oyster Parasite Mikrocytos Mackini, and a Preliminary Phylogenetic Analysis. Dis. Aquat. Org. 2003, 54, 219–227. [Google Scholar] [CrossRef]
- Bower, S.M.; Carnegie, R.B.; Goh, B.; Jones, S.R.; Lowe, G.J.; Mak, M.W. Preferential PCR Amplification of Parasitic Protistan Small Subunit RDNA from Metazoan Tissues. J. Eukaryot. Microbiol. 2004, 51, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.; Pons, M.J.; Herranz, M.; Wakeman, K.C.; del Valle, J.; Vermeij, M.J.A.; Leander, B.S.; Keeling, P.J. Validation of a Universal Set of Primers to Study Animal-associated Microeukaryotic Communities. Env. Microbiol. 2019, 21, 3855–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thu, M.J.; Qiu, Y.; Matsuno, K.; Kajihara, M.; Mori-Kajihara, A.; Omori, R.; Monma, N.; Chiba, K.; Seto, J.; Gokuden, M.; et al. Diversity of Spotted Fever Group Rickettsiae and Their Association with Host Ticks in Japan. Sci. Rep. 2019, 9, 1500. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatov, D.A. Proseq: A Software for Preparation and Evolutionary Analysis of DNA Sequence Data Sets. Mol. Ecol. Notes 2002, 2, 621–624. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.-W.; Richards, T.A. Multiple Marker Parallel Tag Environmental DNA Sequencing Reveals a Highly Complex Eukaryotic Community in Marine Anoxic Water. Mol. Ecol. 2010, 19 (Suppl. S1), 21–31. [Google Scholar] [CrossRef]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.-É.; Carmack, E.C.; Lovejoy, C. Arctic Ocean Microbial Community Structure before and after the 2007 Record Sea Ice Minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Soft. 2015, 67. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive Diversity of RNA Viruses in Australian Ticks. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, T.; Koga, R.; Kikuchi, Y.; Meng, X.-Y.; Fukatsu, T. Wolbachia as a Bacteriocyte-Associated Nutritional Mutualist. Proc. Natl. Acad. Sci. USA 2010, 107, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado-Ferreira, E.; Vizzoni, V.F.; Balsemão-Pires, E.; Moerbeck, L.; Gazeta, G.S.; Piesman, J.; Voloch, C.M.; Soares, C.A.G. Coxiella Symbionts Are Widespread into Hard Ticks. Parasitol. Res. 2016, 115, 4691–4699. [Google Scholar] [CrossRef]
- Pettersson, J.H.-O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the Virome of Ixodes Ricinus Ticks from Northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [Green Version]
- Rio, R.V.M.; Attardo, G.M.; Weiss, B.L. Grandeur Alliances: Symbiont Metabolic Integration and Obligate Arthropod Hematophagy. Trends Parasitol. 2016, 32, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.R.K.; Hemeda, N.F.; Abdelaliem, Y.F. Symbiotic Cellulolytic Bacteria from the Gut of the Subterranean Termite Psammotermes Hypostoma Desneux and Their Role in Cellulose Digestion. Amb. Expr. 2019, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Varm, A.; Kolli, B.K.; Paul, J.; Saxena, S.; König, H. Lignocellulose Degradation by Microorganisms from Termite Hills and Termite Guts: A Survey on the Present State of Art. FEMS Microbiol. Rev. 1994, 15, 9–28. [Google Scholar] [CrossRef]
- Ohkuma, M. Termite Symbiotic Systems: Efficient Bio-Recycling of Lignocellulose. Appl. Microbiol. Biotechnol. 2003, 61, 1–9. [Google Scholar] [CrossRef]
- Koh, C.; Islam, M.N.; Ye, Y.H.; Chotiwan, N.; Graham, B.; Belisle, J.T.; Kouremenos, K.A.; Dayalan, S.; Tull, D.L.; Klatt, S.; et al. Dengue Virus Dominates Lipid Metabolism Modulations in Wolbachia-Coinfected Aedes aegypti. Commun. Biol. 2020, 3, 518. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S RRNA Gene for Designing Universal Eukaryote Specific Primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaaya, G.P.; Hassan, S. Entomogenous Fungi as Promising Biopesticides for Tick Control. Exp. Appl. Acarol. 2000, 24, 913–926. [Google Scholar] [CrossRef]
- Barelli, L.; Moonjely, S.; Behie, S.W.; Bidochka, M.J. Fungi with Multifunctional Lifestyles: Endophytic Insect Pathogenic Fungi. Plant. Mol. Biol 2016, 90, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Rueckert, S.; Betts, E.L.; Tsaousis, A.D. The Symbiotic Spectrum: Where Do the Gregarines Fit? Trends Parasitol. 2019, 35, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Seeber, F.; Steinfelder, S. Recent Advances in Understanding Apicomplexan Parasites. F1000Res 2016, 5, 1369. [Google Scholar] [CrossRef] [Green Version]
- Bollatti, F.; Ceballos, A. Effect of Gregarines (Apicomplexa: Sporozoa) on Survival and Weight Loss of Victorwithius Similis (Arachnida: Pseudoscorpiones). J. Invertebr. Pathol. 2014, 117, 13–18. [Google Scholar] [CrossRef]
- McCray, E.M.; Fay, R.W.; Schoof, H.F. The Bionomics of Lankesteria Culicis and Aedes Aegypti. J. Invertebr. Pathol. 1970, 16, 42–53. [Google Scholar] [CrossRef]
- Lantova, L.; Svobodova, M.; Volf, P. Effects of Psychodiella Sergenti (Apicomplexa, Eugregarinorida) on Its Natural Host Phlebotomus Sergenti (Diptera, Psychodidae). J. Med. Entomol. 2011, 48, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Soghigian, J.; Valsdottir, L.R.; Livdahl, T.P. A Parasite’s Modification of Host Behavior Reduces Predation on Its Host. Ecol. Evol. 2017, 7, 1453–1461. [Google Scholar] [CrossRef]
- Trielli, F.; Chessa, M.G.; Amaroli, A.; Ognibene, M.; Delmonte Corrado, M.U. Effects of Organophosphate Compounds on a Soil Protist, Colpoda Inflata (Ciliophora, Colpodidae). Chemosphere 2006, 65, 1731–1737. [Google Scholar] [CrossRef]
- Cavalier-Smith, T.; Chad, E.E. Sarcomonad Ribosomal RNA Sequences, Rhizopod Phylogeny, and the Origin of Euglyphid Amoebae. Arch. Für Protistenkd. 1997, 147, 227–236. [Google Scholar] [CrossRef]
- Ekelund, F.; Daugbjerg, N.; Fredslund, L. Phylogeny of Heteromita, Cercomonas and Thaumatomonas Based on SSU RDNA Sequences, Including the Description of Neocercomonas Jutlandica Sp. Nov., Gen. Nov. Eur. J. Protistol. 2004, 40, 119–135. [Google Scholar] [CrossRef]
- Hoffmann, A.; Fingerle, V.; Noll, M. Analysis of Tick Surface Decontamination Methods. Microorganisms 2020, 8, 987. [Google Scholar] [CrossRef] [PubMed]
- Kahl, O.; Alidousti, I. Bodies of Liquid Water as a Source of Water Gain for Ixodes ricinus Ticks (Acari: Ixodidae). Exp. Appl. Acarol. 1997, 21, 731–746. [Google Scholar] [CrossRef]
- Meyer-König, A.; Zahler, M.; Gothe, R. Studies on Survival and Water Balance of Unfed Adult Dermacentor marginatus and D. reticulatus Ticks (Acari: Ixodidae). Exp. Appl. Acarol. 2001, 25, 993–1004. [Google Scholar] [CrossRef]
- Maldonado-Ruiz, L.P.; Park, Y.; Zurek, L. Liquid Water Intake of the Lone Star Tick, Amblyomma americanum: Implications for Tick Survival and Management. Sci. Rep. 2020, 10, 6000. [Google Scholar] [CrossRef]
- Portillo, A.; Palomar, A.M.; de Toro, M.; Santibáñez, S.; Santibáñez, P.; Oteo, J.A. Exploring the Bacteriome in Anthropophilic Ticks: To Investigate the Vectors for Diagnosis. PLoS ONE 2019, 14, e0213384. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.E.; Mazel, F.; Lemay, M.A.; Morien, E.; Billy, V.; Kowalewski, M.; Di Fiore, A.; Link, A.; Goldberg, T.L.; Tecot, S.; et al. Biodiversity of Protists and Nematodes in the Wild Nonhuman Primate Gut. ISME J. 2020, 14, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Djurhuus, A.; Closek, C.J.; Kelly, R.P.; Pitz, K.J.; Michisaki, R.P.; Starks, H.A.; Walz, K.R.; Andruszkiewicz, E.A.; Olesin, E.; Hubbard, K.; et al. Environmental DNA Reveals Seasonal Shifts and Potential Interactions in a Marine Community. Nat. Commun. 2020, 11, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, L.; Goubet, A.; Madigou, C.; Pernelle, J.-J.; Palmier, K.; Labadie, K.; Lemainque, A.; Michot, O.; Astoul, L.; Barbier, P.; et al. Colonization Kinetics and Implantation Follow-up of the Sewage Microbiome in an Urban Wastewater Treatment Plant. Sci. Rep. 2020, 10, 11634. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tick ID | Tick Species | Sex | Prefecture | Location | Year |
|---|---|---|---|---|---|
| 66 | I. persulcatus | F | Hokkaido | 42.61, 141.95 | 2013 |
| 210 | I. persulcatus | F | Hokkaido | 42.98, 142.80 | 2013 |
| 258 | I. persulcatus | F | Hokkaido | 43.08, 142.01 | 2013 |
| 467 | I. persulcatus | M | Hokkaido | 43.36, 142.61 | 2013 |
| 932 | I. persulcatus | M | Hokkaido | 43.92, 142.86 | 2014 |
| 933 | I. persulcatus | M | Hokkaido | 43.92, 142.86 | 2014 |
| 2592 | A. testudinarium | M | Shimane | 35.04, 132.68 | 2016 |
| 2874 | A. testudinarium | F | Ehime | 33.35, 132.02 | 2017 |
| 2876 | A. testudinarium | F | Kochi | 33.27, 132.99 | 2017 |
| 3148 | A. testudinarium | F | Ehime | 33.63, 132.56 | 2017 |
| 3149 | A. testudinarium | F | Ehime | 33.63, 132.56 | 2017 |
| 3332 | H. longicornis | F | Hiroshima | 34.49, 132.45 | 2017 |
| 3611 | H. longicornis | F | Chiba | 35.19, 140.25 | 2018 |
| 3620 | H. longicornis | M | Chiba | 35.19, 140.25 | 2018 |
| 3631 | H. longicornis | M | Wakayama | 33.83, 135.30 | 2018 |
| 3643 | H. longicornis | F | Wakayama | 34.37, 135.64 | 2018 |
| 3648 | H. longicornis | M | Kochi | 33.29, 134.17 | 2018 |
| Name | Sequences (5′→3′) | Application | References |
|---|---|---|---|
| Cloning F | CACATCTAAGGAAGGCAGCA | Plasmid preparation | This study |
| Cloning R | CCCTTCCGTCAATTCCTTTA | This study | |
| M13 Primer M4 | GTTTTCCCAGTCACGAC | Plasmid insert sequence identification | Takara Bio Inc. |
| M13 Primer RV | CAGGAAACAGCTATGAC | ||
| TAReuk454FWD1 | CCAGCASCYGCGGTAATTCC | Universal primer sets amplifying V4 region of 18S rDNA | [35] |
| TAReukREV3 | ACTTTCGTTCTTGATYRA | ||
| EUK581F | GTGCCAGCAGCCGCG | UNonMet-PCR (first PCR) | [30] |
| EUK1134R | TTTAAGTTTCAGCCTTGCG | ||
| E572F | CCATCTCATCCCTGCGTGTCTCCGACTCAG | UNonMet-PCR (second PCR) | [36] |
| E1009R | CCTATCCCCTGTGTGCCTTGGCAGTCTCAG | ||
| TickB_PNA | GATCAAWGAAAACATT | Blocking the amplification of tick 18S rDNA | This study |
| TickB_LNA | GATCAAWGAAAACATT 1 | Blocking the amplification of tick 18S rDNA | This study |
| Rank | TickB_PNA-SizeSelect | TickB_LNA-SizeSelect | UNonMet-PCR | |||
|---|---|---|---|---|---|---|
| Higher Category | Genus Level | Higher Category | Genus Level | Higher Category | Genus Level | |
| 1 | Fungi (0.8649%) | Capsicum (0.0703%) | Fungi (1.3424%) | Papiliotrema (0.1527%) | Fungi (6.9371%) | Ascochyta (1.1681%) |
| 2 | Charophyta (0.1386%) | Gregarina (0.0396%) | Charophyta (0.0913%) | Cladosporium (0.0963%) | Ciliophora (0.1170%) | Cladosporium (1.0394%) |
| 3 | Ciliophora (0.0715%) | Mykophagophrys (0.0306%) | Ciliophora (0.0675%) | Ascochyta (0.0913%) | Cercozoa (0.0612%) | Aspergillus (0.7208%) |
| 4 | Apicomplexa (0.0614%) | Ascochyta (0.0230%) | Apicomplexa (0.0133%) | Capsicum (0.0484%) | Apicomplexa (0.0502%) | Didymella (0.2340%) |
| 5 | Cercozoa (0.0310%) | Cladosporium (0.0163%) | Cercozoa (0.0117%) | Vishniacozyma (0.0336%) | Flabellinia (0.0113%) | Penicillium (0.0831%) |
| 6 | Retaria (0.0083%) | Heteromita (0.0135%) | Ochrophyta (0.0049%) | Colpoda (0.0256%) | Dinoflagellata (0.0065%) | Didymosphaeriaceae (0.0686%) |
| 7 | - | Colpoda (0.0127%) | Retaria (0.0020%) | Mykophagophrys (0.0203%) | Cryptomonas (0.0048%) | Roussoella (0.0503%) |
| 8 | - | Spencermartinsiella (0.0048%) | Chlorophyta (0.0019%) | Gregarina (0.0097%) | Didymium (0.0036%) | Gregarina (0.0357%) |
| 9 | - | Meira (0.0048%) | Dinoflagellata (0.0004%) | Heteromita (0.0087%) | Euamoebida (0.0013%) | Heteromita (0.0293%) |
| 10 | - | Aspergillus (0.0043%) | - | Piskurozyma (0.0084%) | Protostelium sp. (0.0012%) | Sorogena (0.0284%) |
| Tick ID | Control | TickB_PNA-AMPure | TickB_PNA-SizeSelect | TickB_LNA-AMPure | TickB_LNA-SizeSelect | UNonMet-PCR |
|---|---|---|---|---|---|---|
| 66 | - | - | - | - | - | - |
| 210 | - | - | - | - | - | Cr |
| 258 | - | - | - | - | - | - |
| 467 | - | - | - | - | - | T |
| 932 | - | - | - | - | - | - |
| 933 | - | - | - | - | - | - |
| 2592 | - | G, A | - | A | - | G, A |
| 2874 | N | G, N | G, N | G, N | G, N | G, EU, N, Co |
| 2876 | - | - | - | - | - | - |
| 3148 | - | - | - | - | - | - |
| 3149 | - | - | - | - | - | - |
| 3332 | - | G | G | G | G | G |
| 3611 | - | - | - | - | - | - |
| 3620 | - | - | - | - | - | - |
| 3631 | - | - | - | - | - | - |
| 3643 | - | - | - | - | - | - |
| 3648 | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taya, Y.; Kinoshita, G.; Mohamed, W.M.A.; Moustafa, M.A.M.; Ogata, S.; Chatanga, E.; Ohari, Y.; Kusakisako, K.; Matsuno, K.; Nonaka, N.; et al. Applications of Blocker Nucleic Acids and Non-Metazoan PCR Improves the Discovery of the Eukaryotic Microbiome in Ticks. Microorganisms 2021, 9, 1051. https://doi.org/10.3390/microorganisms9051051
Taya Y, Kinoshita G, Mohamed WMA, Moustafa MAM, Ogata S, Chatanga E, Ohari Y, Kusakisako K, Matsuno K, Nonaka N, et al. Applications of Blocker Nucleic Acids and Non-Metazoan PCR Improves the Discovery of the Eukaryotic Microbiome in Ticks. Microorganisms. 2021; 9(5):1051. https://doi.org/10.3390/microorganisms9051051
Chicago/Turabian StyleTaya, Yurie, Gohta Kinoshita, Wessam Mohamed Ahmed Mohamed, Mohamed Abdallah Mohamed Moustafa, Shohei Ogata, Elisha Chatanga, Yuma Ohari, Kodai Kusakisako, Keita Matsuno, Nariaki Nonaka, and et al. 2021. "Applications of Blocker Nucleic Acids and Non-Metazoan PCR Improves the Discovery of the Eukaryotic Microbiome in Ticks" Microorganisms 9, no. 5: 1051. https://doi.org/10.3390/microorganisms9051051
APA StyleTaya, Y., Kinoshita, G., Mohamed, W. M. A., Moustafa, M. A. M., Ogata, S., Chatanga, E., Ohari, Y., Kusakisako, K., Matsuno, K., Nonaka, N., & Nakao, R. (2021). Applications of Blocker Nucleic Acids and Non-Metazoan PCR Improves the Discovery of the Eukaryotic Microbiome in Ticks. Microorganisms, 9(5), 1051. https://doi.org/10.3390/microorganisms9051051