
Pathogenic Role of Immune Evasion and Integration of Human Papillomavirus in Oropharyngeal Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
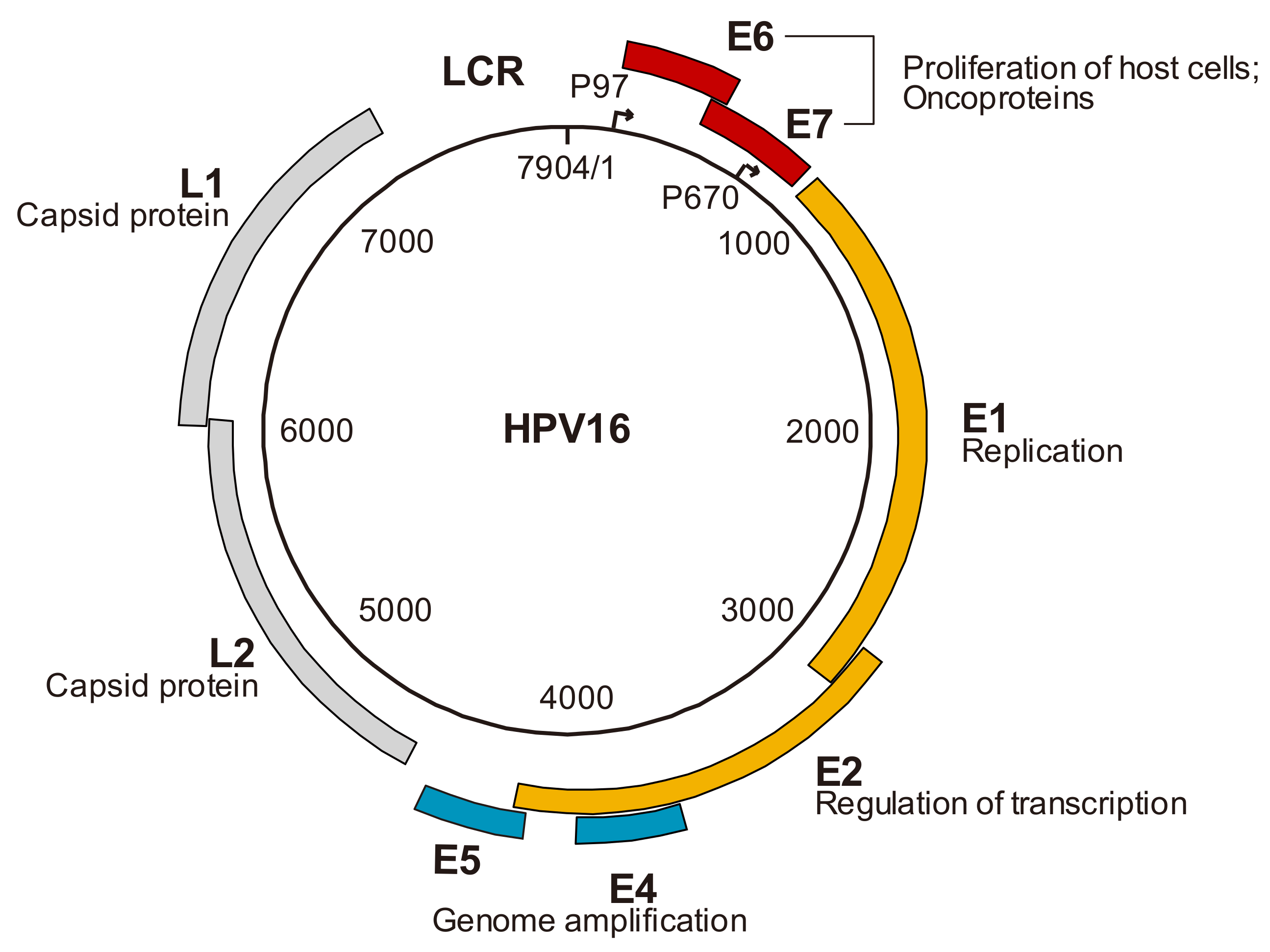
2. The Genomic Structure of HPV and Viral Gene Products
3. HPV Viral Life Cycle
4. Immune Evasion Mechanisms
4.1. Innate Immune System
4.2. Antigen Presentation
4.3. Adaptive Immune System
5. HPV Integration Analysis
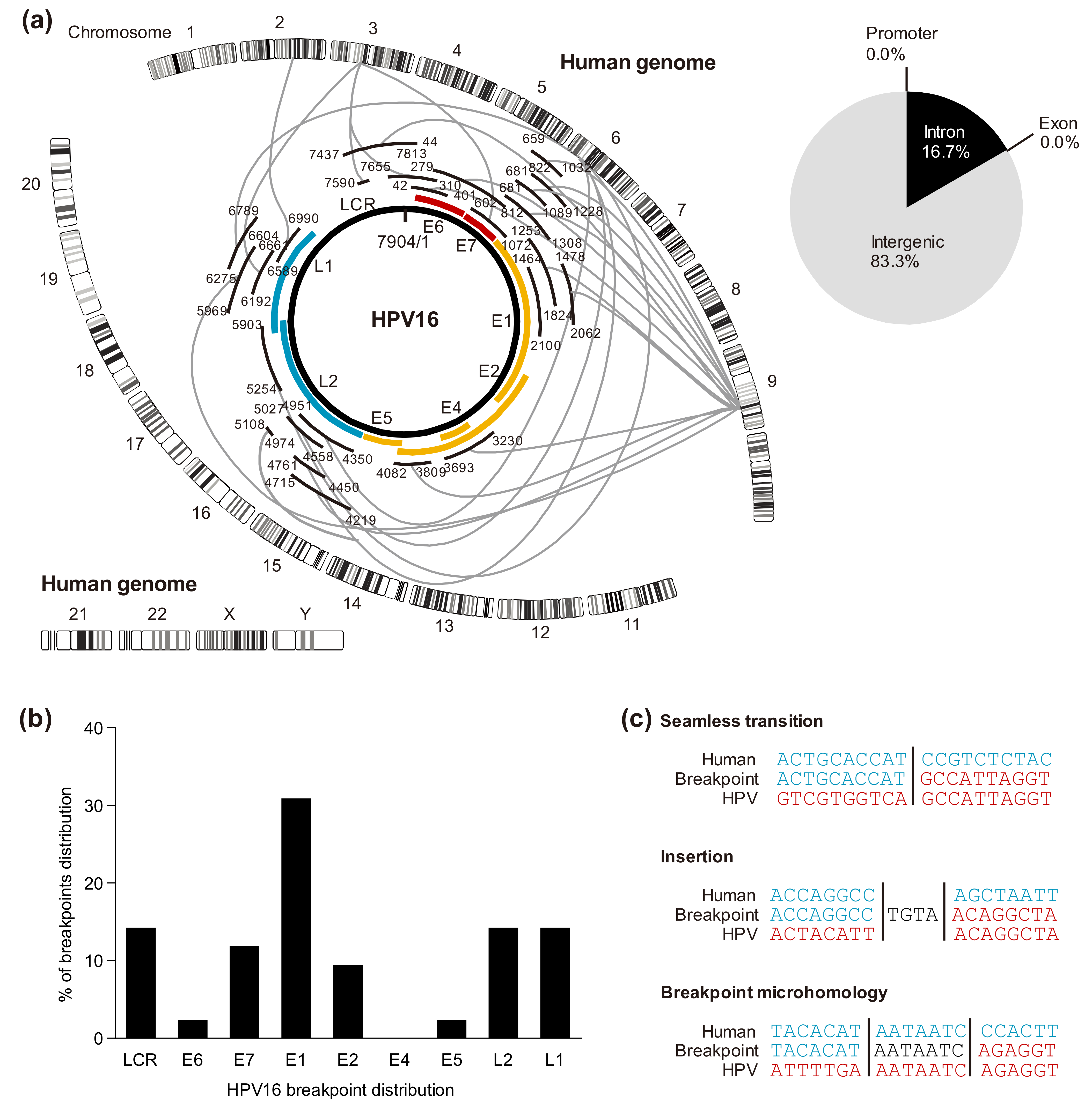
5.1. HPV Genomic Integration Sites in the Host
5.2. Cleavage Sites in the HPV Genome
5.3. Viral-Host Sequences at the Integration Breakpoints
5.4. HPV Integration Types
5.5. Effect of HPV Integration on Cellular Genes
5.6. HPV Integration Affects Epigenetic Alterations
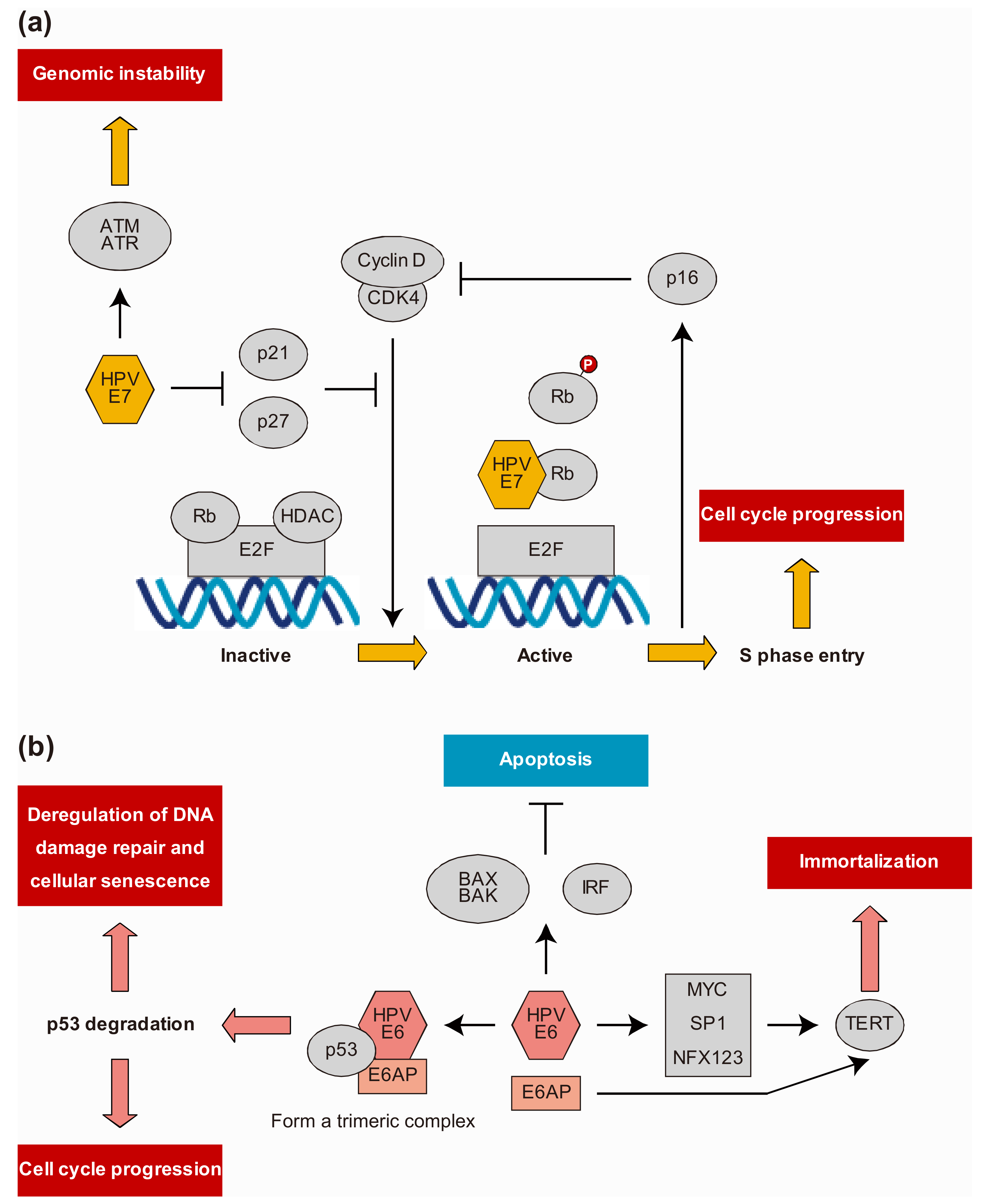
6. HPV Protein-Host Protein Interactions
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Durst, M.; Gissmann, L.; Ikenberg, H.; Hausen, H.Z. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrjänen, K.; Syrjänen, S.; Lamberg, M.; Pyrhönen, S.; Nuutinen, J. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. Int. J. Oral Surg. 1983, 12, 418–424. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; D’Souza, G.; Westra, W.H.; A Forastiere, A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Sano, D.; Oridate, N. The molecular mechanism of human papillomavirus-induced carcinogenesis in head and neck squamous cell carcinoma. Int. J. Clin. Oncol. 2016, 21, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lydiatt, W.M.; Patel, S.G.; O’Sullivan, B.; Brandwein, M.S.; Ridge, J.A.; Migliacci, J.C.; Loomis, A.M.; Shah, J.P. Head and neck cancers-major changes in the American Joint Committee on cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 2012, 41, D571–D578. [Google Scholar] [CrossRef] [Green Version]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; Hausen, H.Z. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausen, H.Z. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, N.; Bosch, F.X.; De Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J.; Griffin, H. Intrabody strategies for the treatment of human papillomavirus-associated disease. Expert Opin. Biol. Ther. 2007, 7, 677–689. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Prim. 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Woodman, C.B.J.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Rautava, J.; Syrjänen, S. Biology of Human Papillomavirus Infections in Head and Neck Carcinogenesis. Head Neck Pathol. 2012, 6, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef] [Green Version]
- Lyford-Pike, S.; Peng, S.; Young, G.D.; Taube, J.M.; Westra, W.H.; Akpeng, B.; Bruno, T.C.; Richmon, J.D.; Wang, H.; Bishop, J.A.; et al. Evidence for a Role of the PD-1:PD-L1 Pathway in Immune Resistance of HPV-Associated Head and Neck Squamous Cell Carcinoma. Cancer Res. 2013, 73, 1733–1741. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M. Immunobiology of HPV and HPV vaccines. Gynecol. Oncol. 2008, 109, S15–S21. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479-480, 146–152. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Ni, G.; Damania, B. Innate Sensing of DNA Virus Genomes. Annu. Rev. Virol. 2018, 5, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Cigno, I.L.; Calati, F.; Albertini, S.; Gariglio, M. Subversion of Host Innate Immunity by Human Papillomavirus Oncoproteins. Pathogens 2020, 9, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, R.; Nakahama, Y.; Nguyen, V.; Espinoza, J.L. The Host-Microbe Interplay in Human Papillomavirus-Induced Carcinogenesis. Microorganisms 2019, 7, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nat. Cell Biol. 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 Expression and Function Is Abolished by the Cervical Cancer-Associated Human Papillomavirus Type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The Human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578. [Google Scholar] [CrossRef] [Green Version]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, C.M.; Doorbar, J.; Nindl, I.; Yoon, H.-S.; Hibma, M.H. Loss of Epidermal Langerhans Cells Occurs in Human Papillomavirus α, γ, and μ but Not β Genus Infections. J. Investig. Dermatol. 2010, 130, 472–480.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guess, J.C.; McCance, D.J. Decreased Migration of Langerhans Precursor-Like Cells in Response to Human Keratinocytes Expressing Human Papillomavirus Type 16 E6/E7 Is Related to Reduced Macrophage Inflammatory Protein-3α Production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional Repression of E-Cadherin by Human Papillomavirus Type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef]
- Bashaw, A.A.; Leggatt, G.R.; Chandra, J.; Tuong, Z.K.; Frazer, I.H. Modulation of antigen presenting cell functions during chronic HPV infection. Papillomavirus Res. 2017, 4, 58–65. [Google Scholar] [CrossRef]
- Kindt, N.; Descamps, G.; Seminerio, I.I.; Bellier, J.J.; Lechien, J.R.; Pottier, C.C.; Larsimont, D.; Journé, F.; O Delvenne, P.; Saussez, S. Langerhans cell number is a strong and independent prognostic factor for head and neck squamous cell carcinomas. Oral Oncol. 2016, 62, 1–10. [Google Scholar] [CrossRef]
- Stanley, M. Immune responses to human papillomavirus. Vaccine 2006, 24, S16–S22. [Google Scholar] [CrossRef]
- Piersma, S.J.; Jordanova, E.S.; Van Poelgeest, M.I.; Kwappenberg, K.M.; Van Der Hulst, J.M.; Drijfhout, J.W.; Melief, C.J.; Kenter, G.G.; Fleuren, G.J.; Offringa, R.; et al. High Number of Intraepithelial CD8+ Tumor-Infiltrating Lymphocytes Is Associated with the Absence of Lymph Node Metastases in Patients with Large Early-Stage Cervical Cancer. Cancer Res. 2007, 67, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Hanna, G.J.; Lizotte, P.; Cavanaugh, M.; Kuo, F.C.; Shivdasani, P.; Frieden, A.; Chau, N.G.; Schoenfeld, J.D.; Lorch, J.H.; Uppaluri, R.; et al. Frameshift events predict anti–PD-1/L1 response in head and neck cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Henderson, S.; Thirdborough, S.M.; Ottensmeier, C.H.; Su, X.; Lechner, M.; Feber, A.; Thomas, G.J.; Fenton, T.R. Human Papillomavirus Drives Tumor Development Throughout the Head and Neck: Improved Prognosis Is Associated With an Immune Response Largely Restricted to the Oropharynx. J. Clin. Oncol. 2016, 34, 4132–4141. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Moerman-Herzog, A.; Nakagawa, M. Early Defensive Mechanisms against Human Papillomavirus Infection. Clin. Vaccine Immunol. 2015, 22, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Şenbabaoğlu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, J.-H.; Lee, J.M.; Choi, J.W.; Jung, D.; Cho, H.; Kang, H.; Hong, M.H.; Heo, S.J.; Kim, S.H.; et al. Molecular subtypes of oropharyngeal cancer show distinct immune microenvironment related with immune checkpoint blockade response. Br. J. Cancer 2020, 122, 1649–1660. [Google Scholar] [CrossRef]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pande, S.; Verma, G.; Hedau, S.; Bhambhani, S.; Kumari, A.; Batra, S.; Basir, S.F.; et al. Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. Indian J. Med. Res. 2014, 139, 531–543. [Google Scholar] [PubMed]
- Mulherkar, R.; Das, P.; Thomas, A.; Kannan, S.; Deodhar, K.; Shrivastava, S.K.; Mahantshetty, U. Human papillomavirus (HPV) genome status & cervical cancer outcome: A retrospective study. Indian J. Med Res. 2015, 142, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Nulton, T.J.; Kim, N.-K.; DiNardo, L.J.; Morgan, I.M.; Windle, B. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol. 2018, 80, 52–55. [Google Scholar] [CrossRef]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Sano, D.; Takahashi, H.; Hyakusoku, H.; Isono, Y.; Shimada, S.; Sawakuma, K.; Takada, K.; Oikawa, R.; Watanabe, Y.; et al. Identification of human papillomavirus (HPV) 16 DNA integration and the ensuing patterns of methylation in HPV-associated head and neck squamous cell carcinoma cell lines. Int. J. Cancer 2017, 140, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [Green Version]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2013, 24, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Meric-Bernstam, F.; Medeiros, L.J.; Weinstein, J.N.; et al. Landscape of DNA Virus Associations across Human Malignant Cancers: Analysis of 3775 Cases Using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Kasperbauer, J.L.; Eckloff, B.W.; Tombers, N.M.; Vasmatzis, G.; Smith, D.I. Mate pair sequencing of oropharyngeal squamous cell carcinomas reveals that HPV integration occurs much less frequently than in cervical cancer. J. Clin. Virol. 2014, 59, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosom. Cancer 2016, 56, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Persing, D.H.; Sarkar, G.; McGovern, R.M.; Gostout, B.S.; Smith, D.I. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 2000, 60, 5916–5921. [Google Scholar] [PubMed]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; I Smith, D. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.Z.; Flam, E.L.; Izumchenko, E.; Danilova, L.V.; Wulf, H.A.; Guo, T.; Singman, D.A.; Afsari, B.; Skaist, A.M.; Considine, M.; et al. Integrated Analysis of Whole-Genome ChIP-Seq and RNA-Seq Data of Primary Head and Neck Tumor Samples Associates HPV Integration Sites with Open Chromatin Marks. Cancer Res. 2017, 77, 6538–6550. [Google Scholar] [CrossRef] [Green Version]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.H.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentzensen, N.; Vinokurova, S.; Doeberitz, M.V.K. Systematic Review of Genomic Integration Sites of Human Papillomavirus Genomes in Epithelial Dysplasia and Invasive Cancer of the Female Lower Genital Tract. Cancer Res. 2004, 64, 3878–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Network, T.C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nat. Cell Biol. 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The Papillomavirus E1 Helicase Activates a Cellular DNA Damage Response in Viral Replication Foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear Accumulation of the Papillomavirus E1 Helicase Blocks S-Phase Progression and Triggers an ATM-Dependent DNA Damage Response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuschenbach, M.; Huebbers, C.U.; Prigge, E.S.; Bermejo, J.L.; Kalteis, M.S.; Preuss, S.F.; Seuthe, I.M.C.; Kolligs, J.; Speel, E.-J.M.; Olthof, N.; et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer 2015, 121, 1966–1976. [Google Scholar] [CrossRef]
- Vartanian, J.-P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for Editing of Human Papillomavirus DNA by APOBEC3 in Benign and Precancerous Lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, S.; Wakae, K.; Wakisaka, N.; Nakanishi, Y.; Ishikawa, K.; Komori, T.; Moriyama-Kita, M.; Endo, K.; Murono, S.; Wang, Z.; et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene 2016, 36, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A Functions as a Restriction Factor of Human Papillomavirus. J. Virol. 2014, 89, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Groves, I.J.; Coleman, N. Human papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburton, A.; Redmond, C.J.; Dooley, K.E.; Fu, H.; Gillison, M.L.; Akagi, K.; Symer, D.E.; Aladjem, M.I.; McBride, A.A. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018, 14, e1007179. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res. 2019, 29, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dooley, K.E.; Warburton, A.; McBride, A.A. Tandemly Integrated HPV16 Can Form a Brd4-Dependent Super-Enhancer-Like Element That Drives Transcription of Viral Oncogenes. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckhardt, M.; Zhang, W.; Gross, A.M.; Von Dollen, J.; Johnson, J.R.; Franks-Skiba, K.E.; Swaney, D.L.; Johnson, T.L.; Jang, G.M.; Shah, P.S.; et al. Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network. Cancer Discov. 2018, 8, 1474–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Chotewutmontri, S.; Wolf, S.; Klös, U.; Schmitz, M.; Dürst, M.; Schwarz, E. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 2013, 8, e66693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.; Patel, M.R.; Cardenas, M.A.; Eberhardt, C.S.; Hudson, W.H.; Obeng, R.C.; Griffith, C.C.; Wang, X.; Chen, Z.G.; Kissick, H.T.; et al. Defining HPV-specific B cell responses in patients with head and neck cancer. Nat. Cell Biol. 2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatano, T.; Sano, D.; Takahashi, H.; Oridate, N. Pathogenic Role of Immune Evasion and Integration of Human Papillomavirus in Oropharyngeal Cancer. Microorganisms 2021, 9, 891. https://doi.org/10.3390/microorganisms9050891
Hatano T, Sano D, Takahashi H, Oridate N. Pathogenic Role of Immune Evasion and Integration of Human Papillomavirus in Oropharyngeal Cancer. Microorganisms. 2021; 9(5):891. https://doi.org/10.3390/microorganisms9050891
Chicago/Turabian StyleHatano, Takashi, Daisuke Sano, Hideaki Takahashi, and Nobuhiko Oridate. 2021. "Pathogenic Role of Immune Evasion and Integration of Human Papillomavirus in Oropharyngeal Cancer" Microorganisms 9, no. 5: 891. https://doi.org/10.3390/microorganisms9050891