
Genomes of Anguillid Herpesvirus 1 Strains Reveal Evolutionary Disparities and Low Genetic Diversity in the Genus Cyprinivirus

 ,
, 
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and AngHV-1 Strains
2.2. AngHV-1 Genome Sequencing
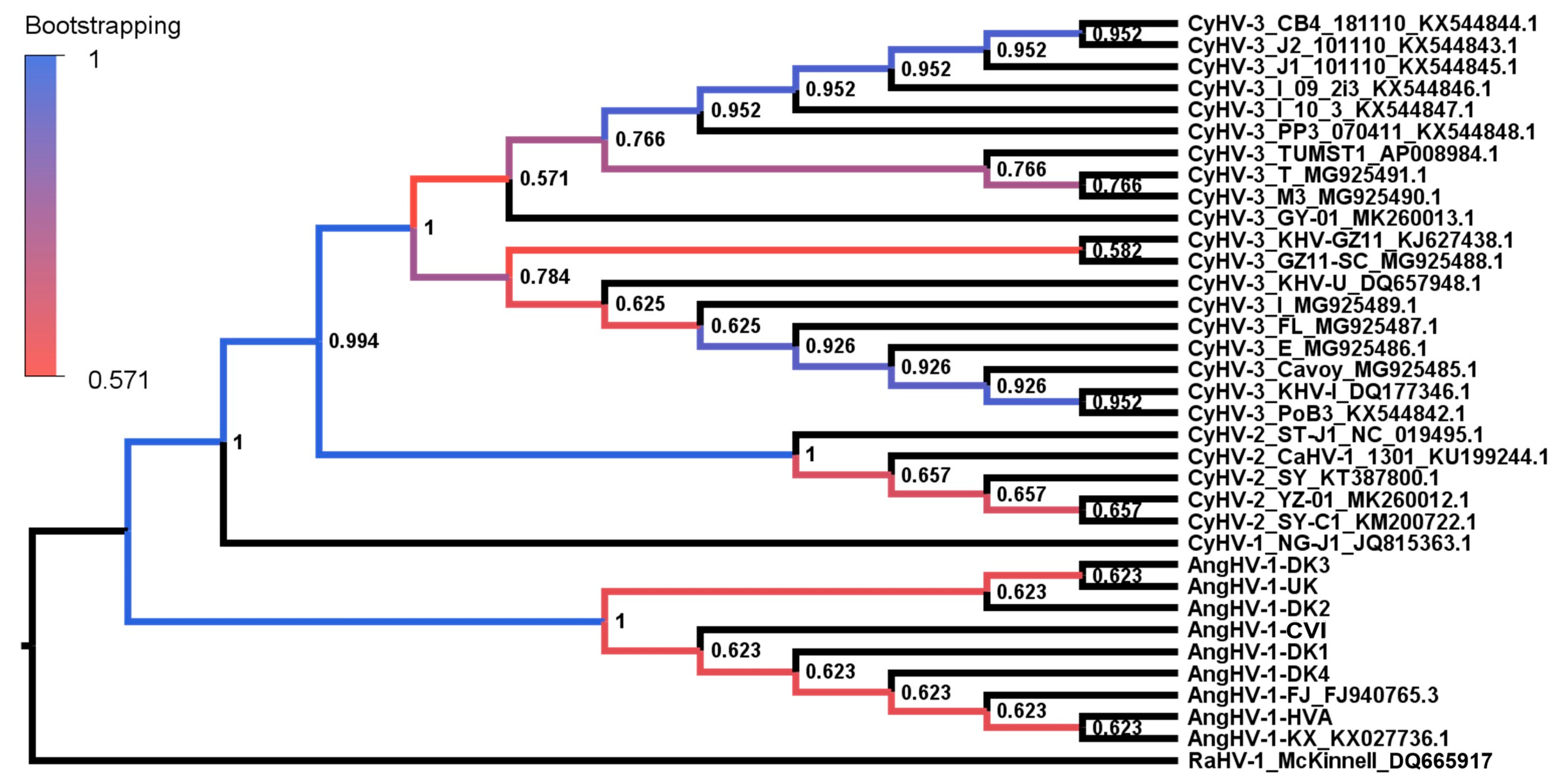
2.3. Phylogenetic Analysis of AngHV-1 Genome Sequences
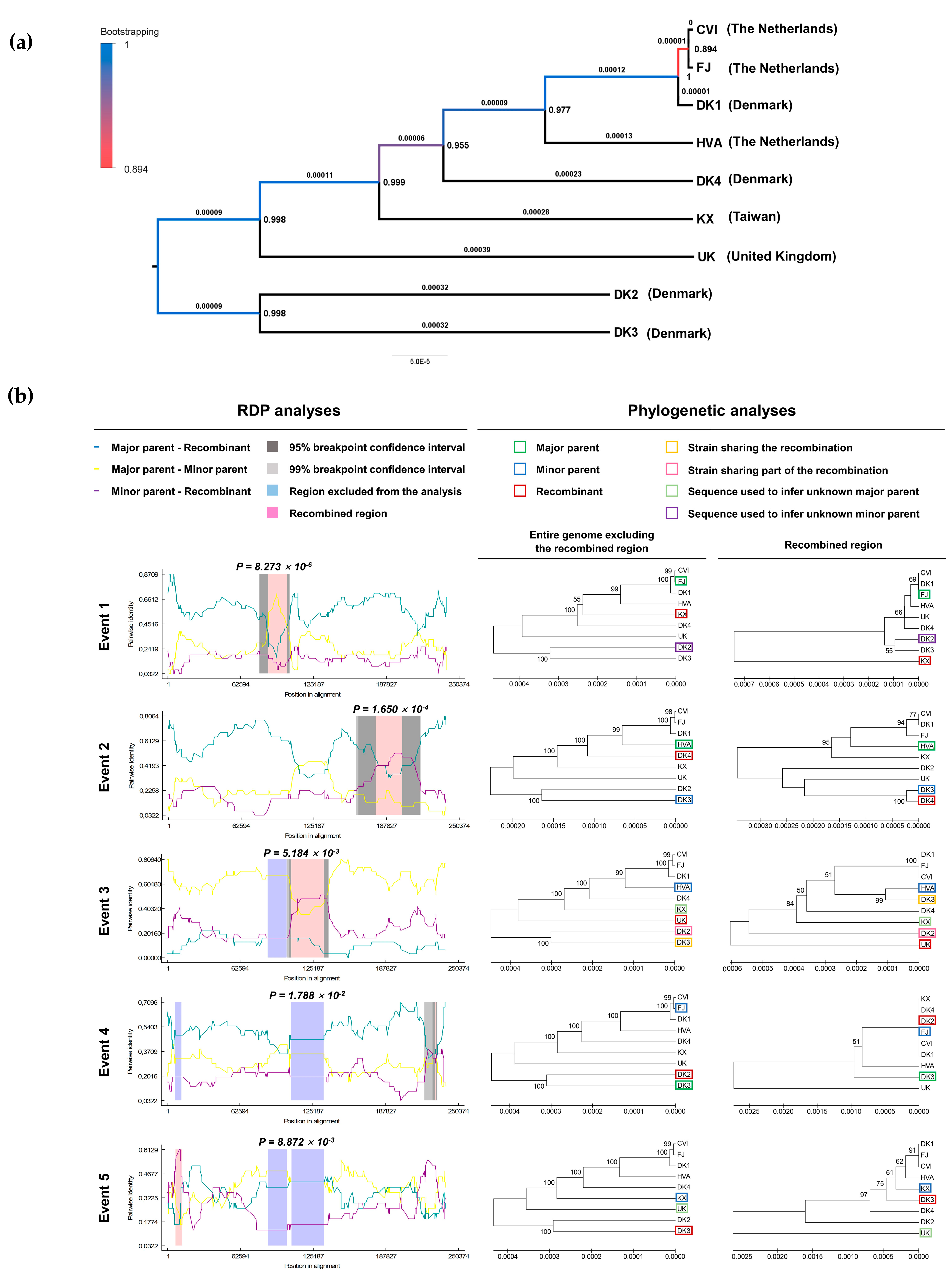
2.4. Recombination Analysis
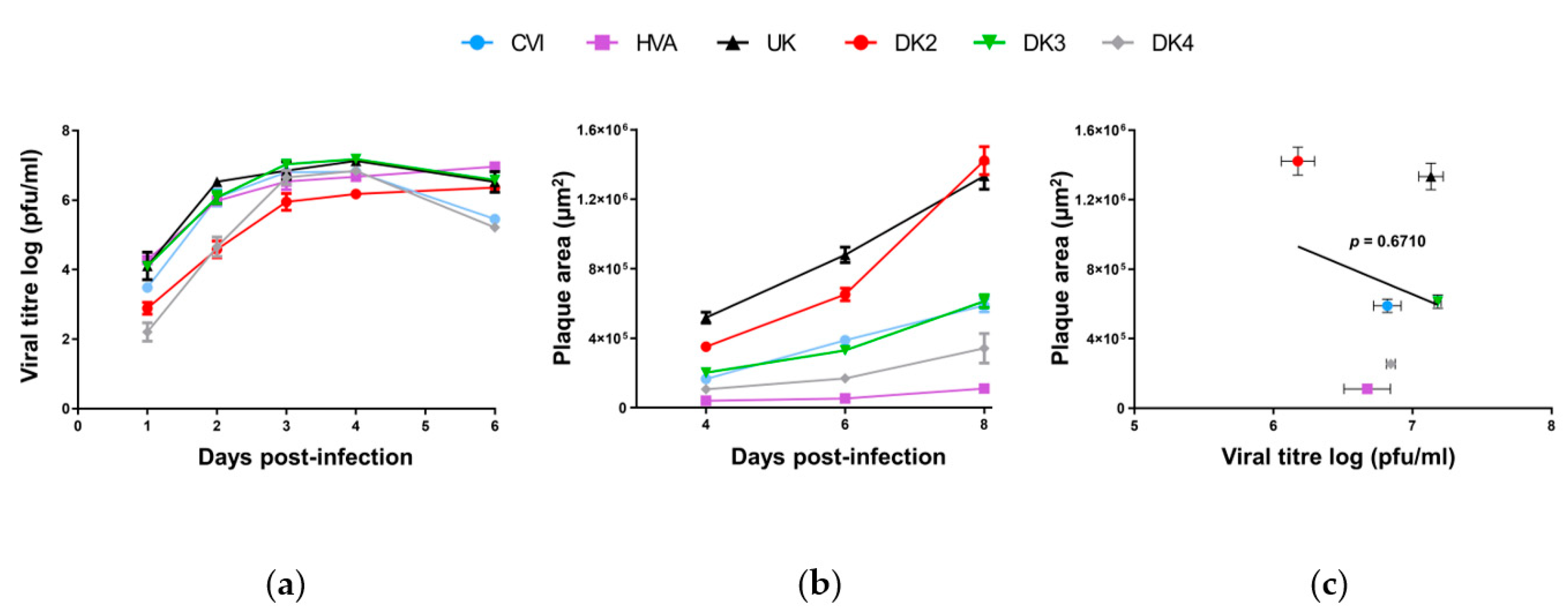
2.5. Viral Growth Assay
2.6. Viral Plaque Area Assay
2.7. Retrieval of Cyprinivirus Core Gene Sequences
2.8. Analysis of Cyprinivirus Core Gene Sequences
2.9. Analysis using Tajima’s Relative Rate Test
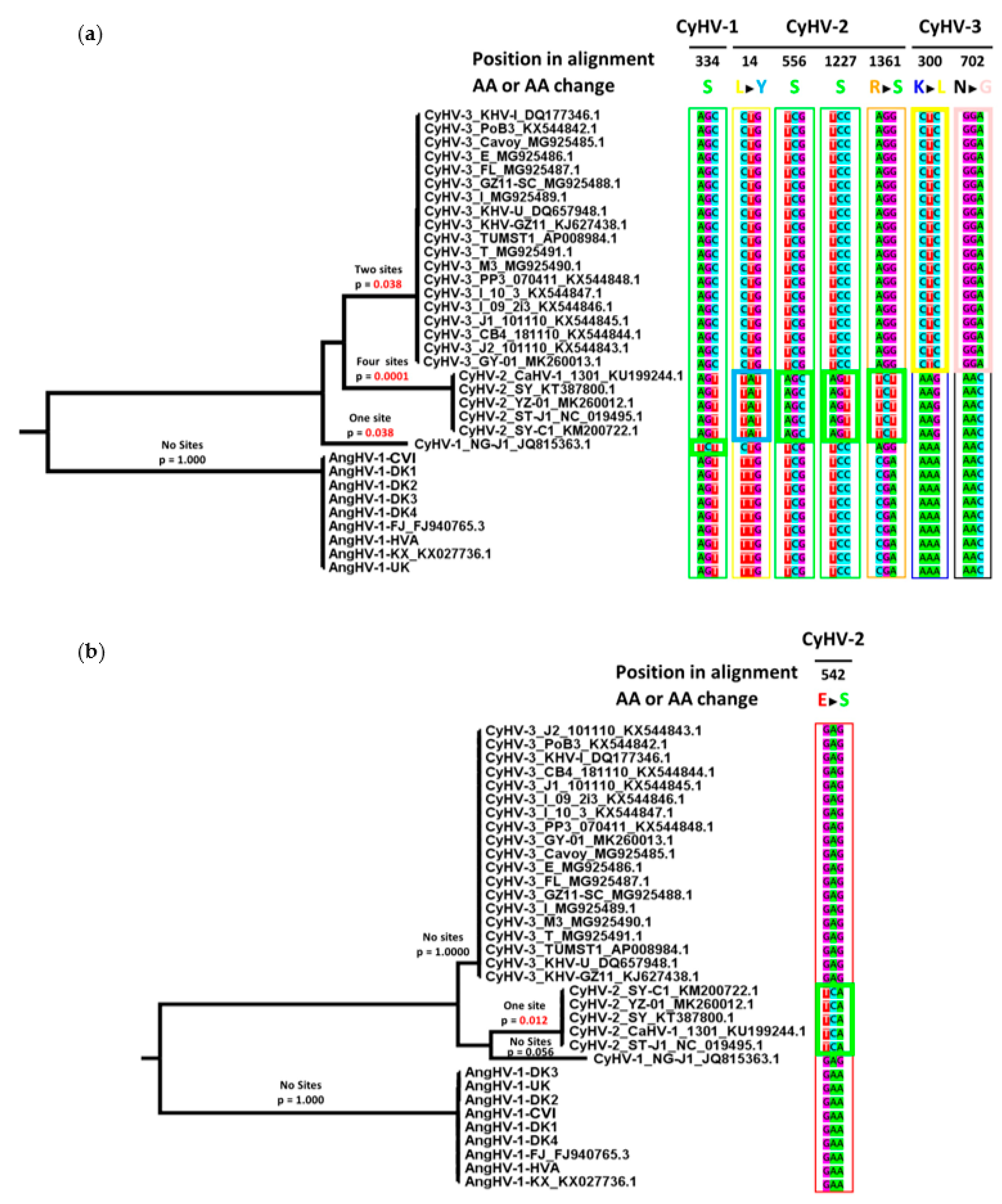
2.10. Analysis of Selective Pressure on Codons
2.11. Estimation of Absolute Divergence Times and Substitution Rates
2.12. Comparison of Species-Level Core Gene Diversity among Cypriniviruses and Members of the Family Herpesviridae
3. Results and Discussion
3.1. Genomic and Biological Comparisons of AngHV-1 Strains
3.2. Evolutionary Comparison of AngHV-1 and Other Cypriniviruses
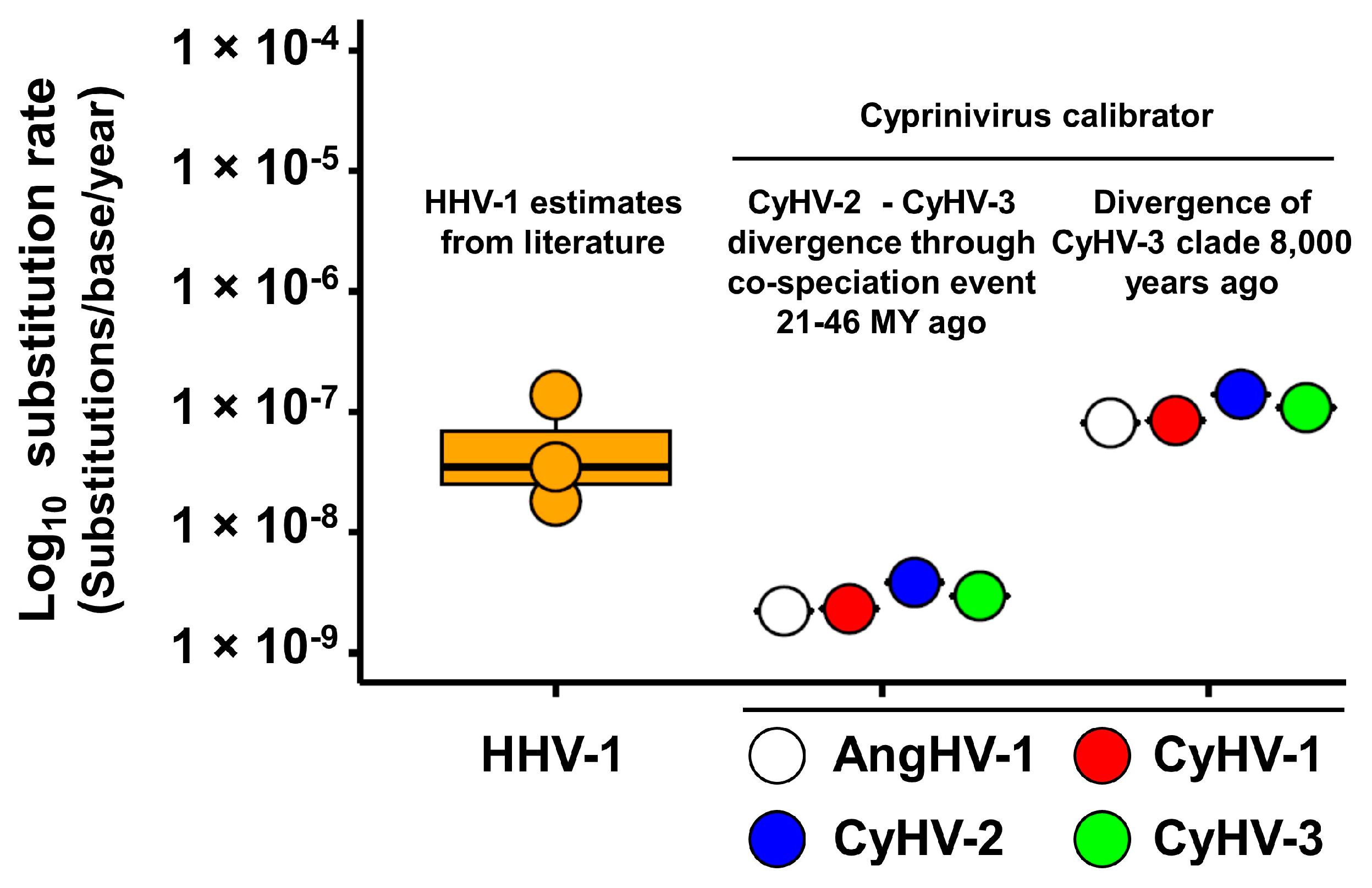
3.3. Absolute Estimates of Substitution Rates and Divergence Times for Cypriniviruses
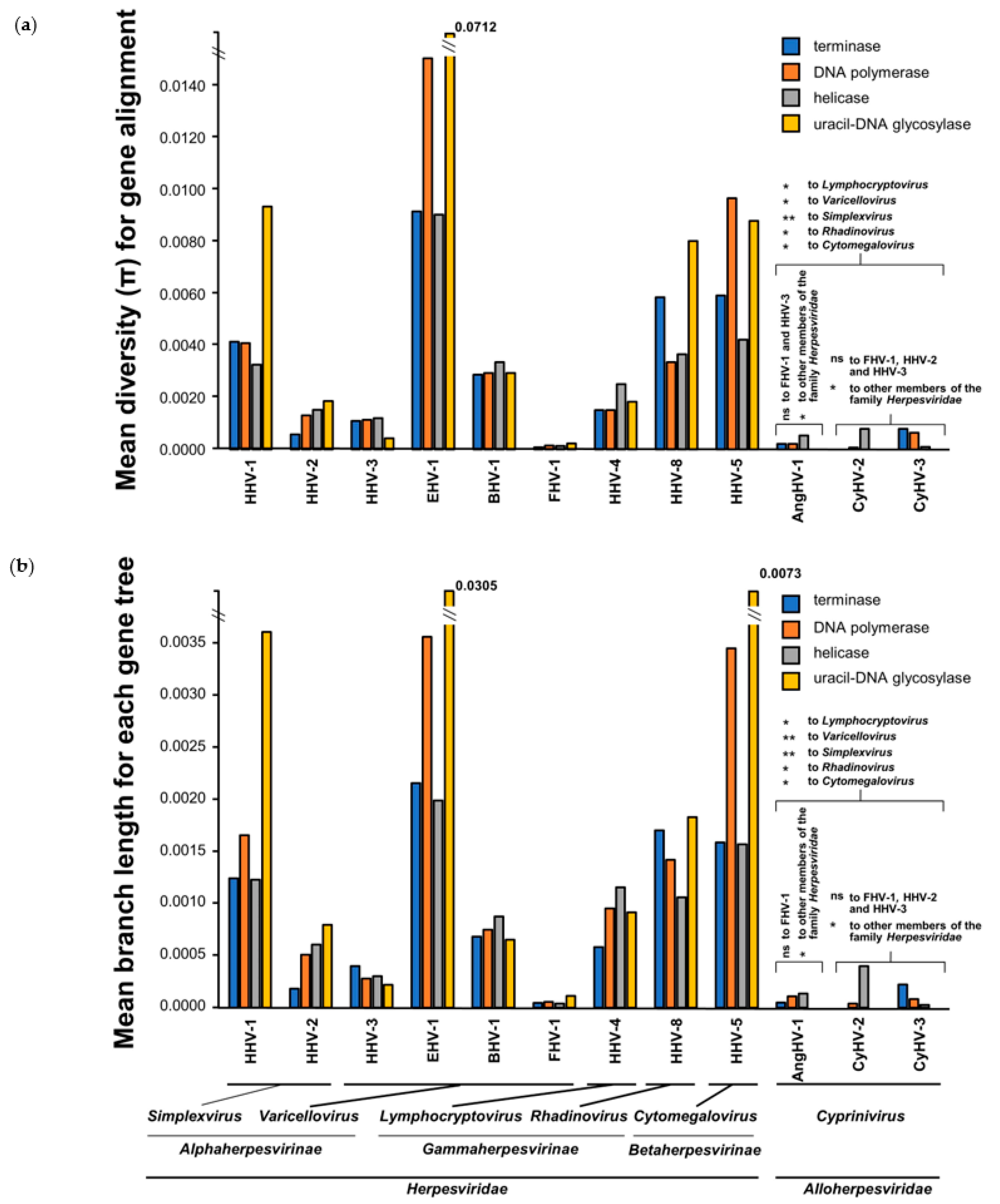
3.4. Comparison of Core Gene Diversity Between Cypriniviruses and Members of the Family Herpesviridae
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The Order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M. Herpesviruses: A Brief Overview. Methods Mol. Biol. 2004, 256, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Roizman, B. The family Herpesviridae: A brief introduction. In Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2479–2499. ISBN 978-0-7817-6060-7. [Google Scholar]
- Pellett, P.E.; Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; Lacoste, V.; Minson, A.C.; Nicholas, J.; Roizman, B.; Studdert, M.J.; et al. Order—Herpesvirales. In Virus Taxonomy; Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012; pp. 99–107. [Google Scholar]
- Boutier, M.; Morvan, L.; Delrez, N.; Origgi, F.; Doszpoly, A.; Vanderplasschen, A. Fish and Amphibian Alloherpesviruses; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 978-0-12-809633-8. [Google Scholar]
- Davison, A.J. Comparative analysis of the genomes. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- McGeoch, D.J.; Davison, A.J.; Dolan, A.; Gatherer, D.; Sevilla-Reyes, E.E. Chapter 20—Molecular evolution of the Herpesvirales. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008; pp. 447–475. ISBN 978-0-12-374153-0. [Google Scholar]
- Mocarski, E.S., Jr. Comparative analysis of herpesvirus-common proteins. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- Davison, A.J.; Kurobe, T.; Gatherer, D.; Cunningham, C.; Korf, I.; Fukuda, H.; Hedrick, R.P.; Waltzek, T.B. Comparative Genomics of Carp Herpesviruses. J. Virol. 2013, 87, 2908–2922. [Google Scholar] [CrossRef] [PubMed]
- Van Beurden, S.J.; Bossers, A.; Voorbergen-Laarman, M.H.A.; Haenen, O.L.M.; Peters, S.; Abma-Henkens, M.H.C.; Peeters, B.P.H.; Rottier, P.J.M.; Engelsma, M.Y. Complete Genome Sequence and Taxonomic Position of Anguillid Herpesvirus 1. J. Gen. Virol. 2010, 91, 880–887. [Google Scholar] [CrossRef]
- Van Beurden, S.J.; Engelsma, M.Y.; Roozenburg, I.; Voorbergen-Laarman, M.A.; van Tulden, P.W.; Kerkhoff, S.; van Nieuwstadt, A.P.; Davidse, A.; Haenen, O.L.M. Viral Diseases of Wild and Farmed European Eel Anguilla Anguilla with Particular Reference to The Netherlands. Dis. Aquat. Organ. 2012, 101, 69–86. [Google Scholar] [CrossRef]
- Van Beurden, S.; Van Engelsma, M. Herpesviruses of fish, amphibians and invertebrates. In Herpesviridae—A Look into This Unique Family of Viruses; IntechOpen: London, UK, 2012. [Google Scholar]
- Chang, P.H.; Pan, Y.H.; Wu, C.M.; Kuo, S.T.; Chung, H.Y. Isolation and Molecular Characterization of Herpesvirus from Cultured European Eels Anguilla Anguilla in Taiwan. Dis. Aquat. Organ. 2002, 50, 111–118. [Google Scholar] [CrossRef]
- Haenen, O.L.M.; Dijkstra, S.G.; van Tulden, P.W.; Davidse, A.; van Nieuwstadt, A.P.; Wagenaar, F.; Wellenberg, G.J. Herpesvirus Anguillae (HVA) Isolations from Disease Outbreaks in Cultured European Eel, Anguilla Anguilla in the Netherlands since 1996. Bull. Eur. Assoc. Fish Pathol. 2002, 22, 247–257. [Google Scholar]
- Sano, M.; Fukuda, H.; Sano, T. Isolation and characterization of a new herpesvirus from eel. In Pathology in Marine Science; Perkins, F.O., Cheng, T.C., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 15–31. ISBN 978-0-12-550755-4. [Google Scholar]
- Bandín, I.; Souto, S.; Cutrín, J.M.; López-Vázquez, C.; Olveira, J.G.; Esteve, C.; Alcaide, E.; Dopazo, C.P. Presence of Viruses in Wild Eels Anguilla Anguilla L., from the Albufera Lake (Spain). J. Fish Dis. 2014, 37, 597–607. [Google Scholar] [CrossRef]
- Dekker, W. The Fractal Geometry of the European Eel Stock. ICES J. Mar. Sci. 2000, 57, 109–121. [Google Scholar] [CrossRef]
- Moriarty, C.; Dekker, W. (Eds.) Management of the European Eel; Irish Fisheries Bulletin, Marine Institute: Galway, Ireland, 1997. [Google Scholar]
- Van Ginneken, V.J.T.; Maes, G.E. The European Eel (Anguilla Anguilla, Linnaeus), Its Lifecycle, Evolution and Reproduction: A Literature Review. Rev. Fish Biol. Fish. 2005, 15, 367–398. [Google Scholar] [CrossRef]
- Békési, L.; Horváth, I.; Kovács-Gayer, E.; Csaba, G. Demonstration of Herpesvirus like Particles in Skin Lesions of European Eel (Anguilla Anguilla). J. Appl. Ichthyol. 1986, 2, 190–192. [Google Scholar] [CrossRef]
- Wen, C.-M.; Liu, P.-C.; Nan, F.-H. Complete Genome Sequence of Herpesvirus Anguillae Strain HVA980811 Isolated in Chiayi, Taiwan. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Chen, S.-N.; Kou, G.-H.; Ueno, Y. A Cell Line Derived from Japanese Eel (Anguilla Japonica) kidney. Proc. Natl. Sci. Counc. Repub. China Part B 1982, 6, 93–100. [Google Scholar]
- Van Beurden, S.J.; Leroy, B.; Wattiez, R.; Haenen, O.L.; Boeren, S.; Vervoort, J.J.; Peeters, B.P.; Rottier, P.J.; Engelsma, M.Y.; Vanderplasschen, A.F. Identification and Localization of the Structural Proteins of Anguillid Herpesvirus 1. Vet. Res. 2011, 42, 105. [Google Scholar] [CrossRef] [PubMed]
- Davidse, A.; Haenen, O.L.M.; Dijkstra, S.G.; van Nieuwstadt, A.P.; van der Vorst, T.J.K.; Wagenaar, F.; Wellenberg, G.J. First Isolation of Herpesvirus of Eel (Herpesvirus Anguillae) in Diseased European Eel (Anguilla Anguilla L.) in Europe. Bull. Eur. Assoc. Fish Pathol. 1999, 19, 137–141. [Google Scholar]
- Boutier, M.; Ronsmans, M.; Ouyang, P.; Fournier, G.; Reschner, A.; Rakus, K.; Wilkie, G.S.; Farnir, F.; Bayrou, C.; Lieffrig, F.; et al. Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging. PLoS Pathog. 2015, 11, e1004690. [Google Scholar] [CrossRef]
- Rakus, K.; Ronsmans, M.; Forlenza, M.; Boutier, M.; Piazzon, M.C.; Jazowiecka-Rakus, J.; Gatherer, D.; Athanasiadis, A.; Farnir, F.; Davison, A.J.; et al. Conserved Fever Pathways across Vertebrates: A Herpesvirus Expressed Decoy TNF-α Receptor Delays Behavioral Fever in Fish. Cell Host Microbe 2017, 21, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of Recombination amongst Aligned Sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Mansai, S.P.; Innan, H. The Power of the Methods for Detecting Interlocus Gene Conversion. Genetics 2010, 184, 517–527. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo Procedure for Assessing Signals in Recombinant Sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Suárez, N.M.; Wilkie, G.S.; Dong, C.; Bergmann, S.; Lee, P.-Y.A.; Davison, A.J.; Vanderplasschen, A.F.C.; Boutier, M. Genomic and Biologic Comparisons of Cyprinid Herpesvirus 3 Strains. Vet. Res. 2018, 49, 40. [Google Scholar] [CrossRef] [PubMed]
- Broeke, J.; Perez, J.M.M.; Pascau, J. Image Processing with ImageJ, 2nd ed.; Packt Publishing: Birmingham, UK, 2015; ISBN 978-1-78588-983-7. [Google Scholar]
- Zeng, X.-T.; Chen, Z.-Y.; Deng, Y.-S.; Gui, J.-F.; Zhang, Q.-Y. Complete Genome Sequence and Architecture of Crucian Carp Carassius Auratus Herpesvirus (CaHV). Arch. Virol. 2016, 161, 3577–3581. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Simple Methods for Testing the Molecular Evolutionary Clock Hypothesis. Genetics 1993, 135, 599–607. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Z. PamlX: A Graphical User Interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef]
- Brandt, A.; Schaefer, I.; Glanz, J.; Schwander, T.; Maraun, M.; Scheu, S.; Bast, J. Effective Purifying Selection in Ancient Asexual Oribatid Mites. Nat. Commun. 2017, 8, 873. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A Visual Tool for Analysis of Selection Using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating Divergence Times in Large Molecular Phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots. 2021. Available online: https://cran.r-project.org/web/packages/ggpubr/ (accessed on 3 May 2021).
- Kolb, A.W.; Ané, C.; Brandt, C.R. Using HSV-1 Genome Phylogenetics to Track Past Human Migrations. PLoS ONE 2013, 8, e76267. [Google Scholar] [CrossRef]
- Norberg, P.; Tyler, S.; Severini, A.; Whitley, R.; Liljeqvist, J.-Å.; Bergström, T. A Genome-Wide Comparative Evolutionary Analysis of Herpes Simplex Virus Type 1 and Varicella Zoster Virus. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Sakaoka, H.; Kurita, K.; Iida, Y.; Takada, S.; Umene, K.; Kim, Y.T.; Ren, C.S.; Nahmias, A.J. Quantitative Analysis of Genomic Polymorphism of Herpes Simplex Virus Type 1 Strains from Six Countries: Studies of Molecular Evolution and Molecular Epidemiology of the Virus. J. Gen. Virol. 1994, 75, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Genetic polymorphism and evolution. In Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000; pp. 231–264. ISBN 978-0-19-513585-5. [Google Scholar]
- Friedman, M. The Use of Ranks to Avoid the Assumption of Normality Implicit in the Analysis of Variance. J. Am. Stat. Assoc. 1937, 32, 675–701. [Google Scholar] [CrossRef]
- Durbin, J. Incomplete Blocks in Ranking Experiments. Br. J. Stat. Psychol. 1951, 4, 85–90. [Google Scholar] [CrossRef]
- Pohlert, T. PMCMR: Calculate Pairwise Multiple Comparisons of Mean Rank Sums; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Delrez, N.; Zhang, H.; Lieffrig, F.; Mélard, C.; Farnir, F.; Boutier, M.; Donohoe, O.; Vanderplasschen, A. European Eel Restocking Programs Based on Wild-Caught Glass Eels: Feasibility of Quarantine Stage Compatible with Implementation of Prophylactic Measures Prior to Scheduled Reintroduction to the Wild. J. Nat. Conserv. 2021, 59, 125933. [Google Scholar] [CrossRef]
- Waltzek, T.B.; Kelley, G.O.; Alfaro, M.E.; Kurobe, T.; Davison, A.J.; Hedrick, R.P. Phylogenetic Relationships in the Family Alloherpesviridae. Dis. Aquat. Organ. 2009, 84, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg-Hua, Y.; Freudenberg, J.; Kluck, N.; Cichon, S.; Propping, P.; Nöthen, M.M. Single Nucleotide Variation Analysis in 65 Candidate Genes for CNS Disorders in a Representative Sample of the European Population. Genome Res. 2003, 13, 2271–2276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J. Rates of Conservative and Radical Nonsynonymous Nucleotide Substitutions in Mammalian Nuclear Genes. J. Mol. Evol. 2000, 50, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, J.P.; Yang, Z. Maximum likelihood methods for detecting adaptive protein evolution. In Statistical Methods in Molecular Evolution; Statistics for Biology and Health; Springer: New York, NY, USA, 2005; pp. 103–124. ISBN 978-0-387-27733-2. [Google Scholar]
- Delport, W.; Scheffler, K.; Seoighe, C. Models of Coding Sequence Evolution. Brief. Bioinform. 2009, 10, 97–109. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.; Frost, S.D.W. Estimating selection pressures on alignments of coding sequences. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Cambridge University Press: Cambridge, UK, 2009; ISBN 978-1-139-47861-8. [Google Scholar]
- Yang, Z. Neutral and adaptive protein evolution. In Computational Molecular Evolution; Oxford University Press: Oxford, UK, 2006; ISBN 978-0-19-172828-0. [Google Scholar]
- Yang, Z.; Nielsen, R. Codon-Substitution Models for Detecting Molecular Adaptation at Individual Sites along Specific Lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef]
- Yang, Z.; dos Reis, M. Statistical Properties of the Branch-Site Test of Positive Selection. Mol. Biol. Evol. 2011, 28, 1217–1228. [Google Scholar] [CrossRef]
- Pitcher, T. Sensory Information and the Organization of Behaviour in a Shoaling Cyprinid Fish. Anim. Behav. 1979, 27, 126–149. [Google Scholar] [CrossRef]
- Smith, J. Social behaviour, homing and migration. In Cyprinid Fishes: Systematics, Biology and Exploitation; Fish & Fisheries Series; Springer: Dordrecht, The Netherlands, 1991; ISBN 978-94-010-5369-3. [Google Scholar]
- Huntingford, F.A.; Andrew, G.; Mackenzie, S.; Morera, D.; Coyle, S.M.; Pilarczyk, M.; Kadri, S. Coping Strategies in a Strongly Schooling Fish, the Common Carp Cyprinus Carpio. J. Fish Biol. 2010, 76, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Malyukina, G.A.; Aleksandryuk, S.P.; Shtefanesku, M. On the Role of Vision in the Schooling Behavior of Minnows (Phoxinus Phoxinus) and the Crucian Carp (Carassius Carassius). Voprosy Ichtiologii 1962, 2, 511–516. [Google Scholar]
- Mann, R.H.K. Growth and production. In Cyprinid Fishes: Systematics, Biology and Exploitation; Fish & Fisheries Series; Springer: Dordrecht, The Netherlands, 1991; ISBN 978-94-010-5369-3. [Google Scholar]
- Pitcher, T.J.; Magurran, A.E.; Winfield, I.J. Fish in Larger Shoals Find Food Faster. Behav. Ecol. Sociobiol. 1982, 10, 149–151. [Google Scholar] [CrossRef]
- Knights, B. Agonistic Behaviour and Growth in the European Eel, Anguilla Anguilla L., in Relation to Warm-Water Aquaculture. J. Fish Biol. 1987, 31, 265–276. [Google Scholar] [CrossRef]
- Burgerhout, E.; Tudorache, C.; Brittijn, S.A.; Palstra, A.P.; Dirks, R.P.; van den Thillart, G.E.E.J.M. Schooling Reduces Energy Consumption in Swimming Male European Eels, Anguilla Anguilla L. J. Exp. Mar. Biol. Ecol. 2013, 448, 66–71. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Otake, T.; Mochioka, N.; Lee, T.-W.; Fricke, H.; Inagaki, T.; Aoyama, J.; Ishikawa, S.; Kimura, S.; Miller, M.J.; et al. Seamounts, New Moon and Eel Spawning: The Search for the Spawning Site of the Japanese Eel. Environ. Biol. Fishes 2003, 66, 221–229. [Google Scholar] [CrossRef]
- Aho, J.; Holopainen, I.J. Batch Spawning of Crucian Carp (Carassius Carassius (L.)) in Mono- and Multispecies Communities. Annales Zoologici Fennici 2000, 37, 101–111. [Google Scholar]
- Beatty, S.J.; Allen, M.G.; Whitty, J.M.; Lymbery, A.J.; Keleher, J.J.; Tweedley, J.R.; Ebner, B.C.; Morgan, D.L. First Evidence of Spawning Migration by Goldfish (Carassius Auratus): Implications for Control of a Globally Invasive Species. Ecol. Freshw. Fish 2017, 26, 444–455. [Google Scholar] [CrossRef]
- Conallin, A.J.; Smith, B.B.; Thwaites, L.A.; Walker, K.F.; Gillanders, B.M. Exploiting the Innate Behaviour of Common Carp, Cyprinus Carpio, to Limit Invasion and Spawning in Wetlands of the River Murray, Australia. Fish. Manag. Ecol. 2016, 23, 431–449. [Google Scholar] [CrossRef]
- Lucas, J.S.; Southgate, P.C.; Tucker, C.S. Aquaculture: Farming Aquatic Animals and Plants; John Wiley & Sons: Hoboken, NJ, USA, 2019; ISBN 978-1-119-23086-1. [Google Scholar]
- Sarkar, A.; Upadhyay, B. Histochemical Detection of Steroid Synthesizing Cells in the Testes of Goldfish, Carassius Auratus, during the Annual Reproductive Cycle. Biotech. Histochem. 2014, 89, 114–120. [Google Scholar] [CrossRef]
- Şaşı, H. The Length and Weight Relations of Some Reproduction Characteristics of Prussian Carp, Carassius Gibelio (Bloch, 1782) in the South Aegean Region (Aydın-Turkey). Turk. J. Fish. Aquat. Sci. 2008, 8, 87–92. [Google Scholar]
- Sivakumaran, K.P.; Brown, P.; Stoessel, D.; Giles, A. Maturation and Reproductive Biology of Female Wild Carp, Cyprinus Carpio, in Victoria, Australia. Environ. Biol. Fishes 2003, 68, 321–332. [Google Scholar] [CrossRef]
- Teletchea, F.; Fostier, A.; Kamler, E.; Gardeur, J.-N.; Le Bail, P.-Y.; Jalabert, B.; Fontaine, P. Comparative Analysis of Reproductive Traits in 65 Freshwater Fish Species: Application to the Domestication of New Fish Species. Rev. Fish Biol. Fish. 2009, 19, 403–430. [Google Scholar] [CrossRef]
- Berry, L.; Brookes, D.; Walker, B. The Problem of the Migration of the European Eel (Anguilla Anguilla). Sci. Prog. 1972, 60, 465–485. [Google Scholar]
- Cresci, A. A Comprehensive Hypothesis on the Migration of European Glass Eels (Anguilla Anguilla). Biol. Rev. 2020, 95, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.; Dishon, A.; Kotler, M. Herpesviruses That Infect Fish. Viruses 2011, 3, 2160–2191. [Google Scholar] [CrossRef]
- Rakus, K.; Ronsmans, M.; Vanderplasschen, A. Behavioral Fever in Ectothermic Vertebrates. Dev. Comp. Immunol. 2017, 66, 84–91. [Google Scholar] [CrossRef]
- Gilad, O.; Yun, S.; Zagmutt-Vergara, F.J.; Leutenegger, C.M.; Bercovier, H.; Hedrick, R.P. Concentrations of a Koi Herpesvirus (KHV) in Tissues of Experimentally Infected Cyprinus Carpio Koi as Assessed by Real-Time TaqMan PCR. Dis. Aquat. Organ. 2004, 60, 179–187. [Google Scholar] [CrossRef]
- Gilad, O.; Yun, S.; Adkison, M.A.; Way, K.; Willits, N.H.; Bercovier, H.; Hedrick, R.P. Molecular Comparison of Isolates of an Emerging Fish Pathogen, Koi Herpesvirus, and the Effect of Water Temperature on Mortality of Experimentally Infected Koi. J. Gen. Virol. 2003, 84, 2661–2667. [Google Scholar] [CrossRef] [PubMed]
- Groff, J.M.; LaPatra, S.E.; Munn, R.J.; Zin, J.G. A Viral Epizootic in Cultured Populations of Juvenile Goldfish Due to a Putative Herpesvirus Etiology. J. Vet. Diagn. Investig. 1998, 10, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, K.R.; Bateman, K.; Bayley, A.; Feist, S.W.; Hulland, J.; Longshaw, C.; Stone, D.; Woolford, G.; Way, K. Isolation of a Cyprinid Herpesvirus 2 from Goldfish, Carassius Auratus (L.), in the UK. J. Fish Dis. 2007, 30, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Moriwake, M.; Sano, T. Herpesvirus Cyprini: Thermal Effects on Pathogenicity and Oncogenicity. Fish Pathol. 1993, 28, 171–175. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Dolan, A.; Ralph, A.C. Toward a Comprehensive Phylogeny for Mammalian and Avian Herpesviruses. J. Virol. 2000, 74, 10401–10406. [Google Scholar] [CrossRef]
- Aoki, T.; Hirono, I.; Kurokawa, K.; Fukuda, H.; Nahary, R.; Eldar, A.; Davison, A.J.; Waltzek, T.B.; Bercovier, H.; Hedrick, R.P. Genome Sequences of Three Koi Herpesvirus Isolates Representing the Expanding Distribution of an Emerging Disease Threatening Koi and Common Carp Worldwide. J. Virol. 2007, 81, 5058–5065. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Cook, S.; Dolan, A.; Jamieson, F.E.; Telford, E.A. Molecular Phylogeny and Evolutionary Timescale for the Family of Mammalian Herpesviruses. J. Mol. Biol. 1995, 247, 443–458. [Google Scholar] [CrossRef]
- Kennedy, D.A.; Kurath, G.; Brito, I.L.; Purcell, M.K.; Read, A.F.; Winton, J.R.; Wargo, A.R. Potential Drivers of Virulence Evolution in Aquaculture. Evol. Appl. 2016, 9, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.G. Epidemiology of the Spread of Viral Diseases under Aquaculture. Curr. Opin. Virol. 2013, 3, 74–78. [Google Scholar] [CrossRef]
- Murray, A.G.; Peeler, E.J. A Framework for Understanding the Potential for Emerging Diseases in Aquaculture. Prev. Vet. Med. 2005, 67, 223–235. [Google Scholar] [CrossRef]
- Pulkkinen, K.; Suomalainen, L.-R.; Read, A.F.; Ebert, D.; Rintamäki, P.; Valtonen, E.T. Intensive Fish Farming and the Evolution of Pathogen Virulence: The Case of Columnaris Disease in Finland. Proc. R. Soc. B Biol. Sci. 2010, 277, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, L.-R.; Ketola, T.; Laanto, E.; Kinnula, H.; Bamford, J.K.H.; Penttinen, R.; Mappes, J. Intensive Aquaculture Selects for Increased Virulence and Interference Competition in Bacteria. Proc. Biol. Sci. 2016, 283, 20153069. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Hudson, M.J.; Uchiyama, J.; Makibayashi, K.; Zhang, J. Common Carp Aquaculture in Neolithic China Dates Back 8000 Years. Nat. Ecol. Evol. 2019, 3, 1415–1418. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Time Dependency of Foamy Virus Evolutionary Rate Estimates. BMC Evol. Biol. 2015, 15. [Google Scholar] [CrossRef]
- Duchêne, S.; Holmes, E.C.; Ho, S.Y.W. Analyses of Evolutionary Dynamics in Viruses Are Hindered by a Time-Dependent Bias in Rate Estimates. Proc. R. Soc. B Biol. Sci. 2014, 281. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Duchêne, S.; Molak, M.; Shapiro, B. Time-Dependent Estimates of Molecular Evolutionary Rates: Evidence and Causes. Mol. Ecol. 2015, 24, 6007–6012. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of War—Host Adaptation and Its Constraints on Virus Evolution. Nat. Rev. Microbiol. 2019, 17, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Van Tuinen, M.; Torres, C.R. Potential for Bias and Low Precision in Molecular Divergence Time Estimation of the Canopy of Life: An Example from Aquatic Bird Families. Front. Genet. 2015, 6. [Google Scholar] [CrossRef]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef]
- Forni, D.; Pontremoli, C.; Clerici, M.; Pozzoli, U.; Cagliani, R.; Sironi, M. Recent Out-of-Africa Migration of Human Herpes Simplex Viruses. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-Dependent Rates of Molecular Evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef]
- Burrel, S.; Désiré, N.; Marlet, J.; Dacheux, L.; Seang, S.; Caumes, E.; Bourhy, H.; Agut, H.; Boutolleau, D. Genetic Diversity within Alphaherpesviruses: Characterization of a Novel Variant of Herpes Simplex Virus 2. J. Virol. 2015, 89, 12273–12283. [Google Scholar] [CrossRef]
- Lamers, S.L.; Newman, R.M.; Laeyendecker, O.; Tobian, A.A.R.; Colgrove, R.C.; Ray, S.C.; Koelle, D.M.; Cohen, J.; Knipe, D.M.; Quinn, T.C. Global Diversity within and between Human Herpesvirus 1 and 2 Glycoproteins. J. Virol. 2015, 89, 8206–8218. [Google Scholar] [CrossRef]
- McGeoch, D.J. Lineages of Varicella-Zoster Virus. J. Gen. Virol. 2009, 90, 963–969. [Google Scholar] [CrossRef]
- Vaz, P.K.; Job, N.; Horsington, J.; Ficorilli, N.; Studdert, M.J.; Hartley, C.A.; Gilkerson, J.R.; Browning, G.F.; Devlin, J.M. Low Genetic Diversity among Historical and Contemporary Clinical Isolates of Felid Herpesvirus 1. BMC Genom. 2016, 17. [Google Scholar] [CrossRef]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the Herpes Simplex Virus-1 DNA Polymerase and Uracil DNA Glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef] [PubMed]
- Su, M.-T.; Liu, I.-H.; Wu, C.-W.; Chang, S.-M.; Tsai, C.-H.; Yang, P.-W.; Chuang, Y.-C.; Lee, C.-P.; Chen, M.-R. Uracil DNA Glycosylase BKRF3 Contributes to Epstein-Barr Virus DNA Replication through Physical Interactions with Proteins in Viral DNA Replication Complex. J. Virol. 2014, 88, 8883–8899. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Savva, R. The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design. Microorganisms 2020, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Haenen, O.L.M.; Way, K.; Bergmann, S.M.; Ariel, E. The Emergence of Koi Herpesvirus and Its Significance to European Aquaculture. Bull. Eur. Assoc. Fish Pathol. 2004, 24, 293–307. [Google Scholar]
- Ito, T.; Kurita, J.; Haenen, O.L.M. Importation of CyHV-2-Infected Goldfish into the Netherlands. Dis. Aquat. Organ. 2017, 126, 51–62. [Google Scholar] [CrossRef]
- Kempter, J.; Hofsoe, P.; Panicz, R.; Bergmann, S. First Detection of Anguillid Herpesvirus 1 (AngHV1) in European Eel (Anguilla Anguilla) and Imported American Eel (Anguilla Rostrata) in Poland. Bull. Eur. Assoc. Fish Pathol. 2014, 34, 87–94. [Google Scholar]
- Kullmann, B.; Adamek, M.; Steinhagen, D.; Thiel, R. Anthropogenic Spreading of Anguillid Herpesvirus 1 by Stocking of Infected Farmed European Eels, Anguilla Anguilla (L.), in the Schlei Fjord in Northern Germany. J. Fish Dis. 2017, 40, 1695–1706. [Google Scholar] [CrossRef]
- Panicz, R.; Sadowski, J.; Eljasik, P. Detection of Cyprinid Herpesvirus 2 (CyHV-2) in Symptomatic Ornamental Types of Goldfish (Carassius Auratus) and Asymptomatic Common Carp (Cyprinus Carpio) Reared in Warm-Water Cage Culture. Aquaculture 2019. [Google Scholar] [CrossRef]
- Pearson, H. Carp Virus Crisis Prompts Moves to Avert Global Spread. Nature 2004, 427, 577. [Google Scholar] [CrossRef] [PubMed]
- Pokorova, D.; Vesely, T.; Piackova, V. Current Knowledge on Koi Herpesvirus—A Review. Veterinární Med. 2005, 50, 139–147. [Google Scholar] [CrossRef]
- Boutier, M.; Donohoe, O.; Kopf, R.K.; Humphries, P.; Becker, J.A.; Marshall, J.; Vanderplasschen, A. Biocontrol of Carp: The Australian Plan Does Not Stand Up to a Rational Analysis of Safety and Efficacy. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kopf, R.K.; Boutier, M.; Finlayson, C.M.; Hodges, K.; Humphries, P.; King, A.; Kingsford, R.T.; Marshall, J.; McGinness, H.M.; Thresher, R.; et al. Biocontrol in Australia: Can a Carp Herpesvirus (CyHV-3) Deliver Safe and Effective Ecological Restoration? Biol. Invasions 2019, 21, 1857–1870. [Google Scholar] [CrossRef]
- Marshall, J.; Davison, A.J.; Kopf, R.K.; Boutier, M.; Stevenson, P.; Vanderplasschen, A. Biocontrol of Invasive Carp: Risks Abound. Science 2018, 359, 877. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short Name Used in This Study | Original Name | Geographic Origin | Host | GenBank Accession Number | References |
|---|---|---|---|---|---|
| CVI 1 | CVI NL 500138 | The Netherlands | Anguilla anguilla | MW580849 | [10] |
| HVA 1 | HVA486123 | MW580854 | [14,24] | ||
| UK 1 | UK80 | United Kingdom | MW580855 | - | |
| DK1 1 | DK-200249 | Denmark | MW580851 | - | |
| DK2 1 | DK-2008-50-66-1 | MW580850 | - | ||
| DK3 1 | DK-205223-2 | MW580852 | - | ||
| DK4 1 | DK-206116-1 | MW580853 | - | ||
| FJ | The Netherlands | FJ940765.3 | [10] | ||
| KX | Taiwan | Anguilla japonica | KX027736.1 | [21] |
| Strain | ORF 1 | |||||
|---|---|---|---|---|---|---|
| CVI | 25 3 | |||||
| DK1 | ||||||
| HVA | 102 | |||||
| DK4 | (15) 2 | (25) 3 | (76) 3 | 102 | 112 4 | |
| KX | 102 | 112 5 | ||||
| UK | 102 | 112 5 | ||||
| DK2 | (15) 2 | (53) | (76) 3 | 102 | 112 4 | |
| DK3 | 102 | 112 4 | ||||
| Gene | Viral Species | Position in Alignment 1 | AA Sites Under Positive Selection (Location in Relevant Species) | Bayes Posterior Probability 3 |
|---|---|---|---|---|
| DNA polymerase | CyHV-1 | 334 | 314S 2 | 0.963 |
| CyHV-2 | 14 | 14Y | 0.964 | |
| 556 | 510S 2 | 0.962 | ||
| 1227 | 1150S 2 | 0.966 | ||
| 1361 | 1282S | 0.965 | ||
| CyHV-3 | 300 | 282L | 0.976 | |
| 702 | 656G | 0.951 | ||
| helicase | CyHV-2 | 542 | 508S | 0.959 |
| Calibration Hypotheses 1 | Species | AA | Codon (Position 1 + 2) | ||
|---|---|---|---|---|---|
| Divergence Time (In Years) and Confidence Intervals | Substitution Rate (Per Site Per Year) 2 | Divergence Time (In Years) and Confidence Intervals | Substitution Rate (Per Base Per Year) 2 | ||
| Divergence of CyHV-2 and CyHV-3 21–46 MYA | CyHV-1 | N/A | 3.48 × 10−9 | N/A | 2.33 × 10−9 |
| CyHV-2 | 26,735 (16,980–42,092) | 5.96 × 10−9 | 9348 (7420–11,777) | 3.85 × 10−9 | |
| CyHV-3 | 181,183 (91,633–358,249) | 4.81 × 10−9 | 292,158 (233,744–365,171) | 2.97 × 10−9 | |
| AngHV-1 | 23,341 (11,091–49,123) | 3.22 × 10−9 | 16,657 (8138–34,096) | 2.23 × 10−9 | |
| Mean | N/A | 4.37 × 10−9 | N/A | 2.84 × 10−9 | |
| Divergence of CyHV-3 clade 8000 years ago | CyHV-1 | N/A | 7.87 × 10−8 | N/A | 8.47 × 10−8 |
| CyHV-2 | 521 (420–647) | 1.37 × 10−7 | 256 (139–470) | 1.40 × 10−7 | |
| CyHV-3 | 8000 (6400–10,000) | 1.09 × 10−7 | 8000 (6570-9741) | 1.08 × 10−7 | |
| AngHV-1 | 1032 (918–1160) | 7.29 × 10−8 | 456 (223–934) | 8.11 × 10−8 | |
| Mean | N/A | 9.93 × 10−8 | N/A | 1.04 × 10−7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donohoe, O.; Zhang, H.; Delrez, N.; Gao, Y.; Suárez, N.M.; Davison, A.J.; Vanderplasschen, A. Genomes of Anguillid Herpesvirus 1 Strains Reveal Evolutionary Disparities and Low Genetic Diversity in the Genus Cyprinivirus. Microorganisms 2021, 9, 998. https://doi.org/10.3390/microorganisms9050998
Donohoe O, Zhang H, Delrez N, Gao Y, Suárez NM, Davison AJ, Vanderplasschen A. Genomes of Anguillid Herpesvirus 1 Strains Reveal Evolutionary Disparities and Low Genetic Diversity in the Genus Cyprinivirus. Microorganisms. 2021; 9(5):998. https://doi.org/10.3390/microorganisms9050998
Chicago/Turabian StyleDonohoe, Owen, Haiyan Zhang, Natacha Delrez, Yuan Gao, Nicolás M. Suárez, Andrew J. Davison, and Alain Vanderplasschen. 2021. "Genomes of Anguillid Herpesvirus 1 Strains Reveal Evolutionary Disparities and Low Genetic Diversity in the Genus Cyprinivirus" Microorganisms 9, no. 5: 998. https://doi.org/10.3390/microorganisms9050998
APA StyleDonohoe, O., Zhang, H., Delrez, N., Gao, Y., Suárez, N. M., Davison, A. J., & Vanderplasschen, A. (2021). Genomes of Anguillid Herpesvirus 1 Strains Reveal Evolutionary Disparities and Low Genetic Diversity in the Genus Cyprinivirus. Microorganisms, 9(5), 998. https://doi.org/10.3390/microorganisms9050998