Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Processing and Sequencing
2.2. Sequence Read Processing
2.3. Published Sequences
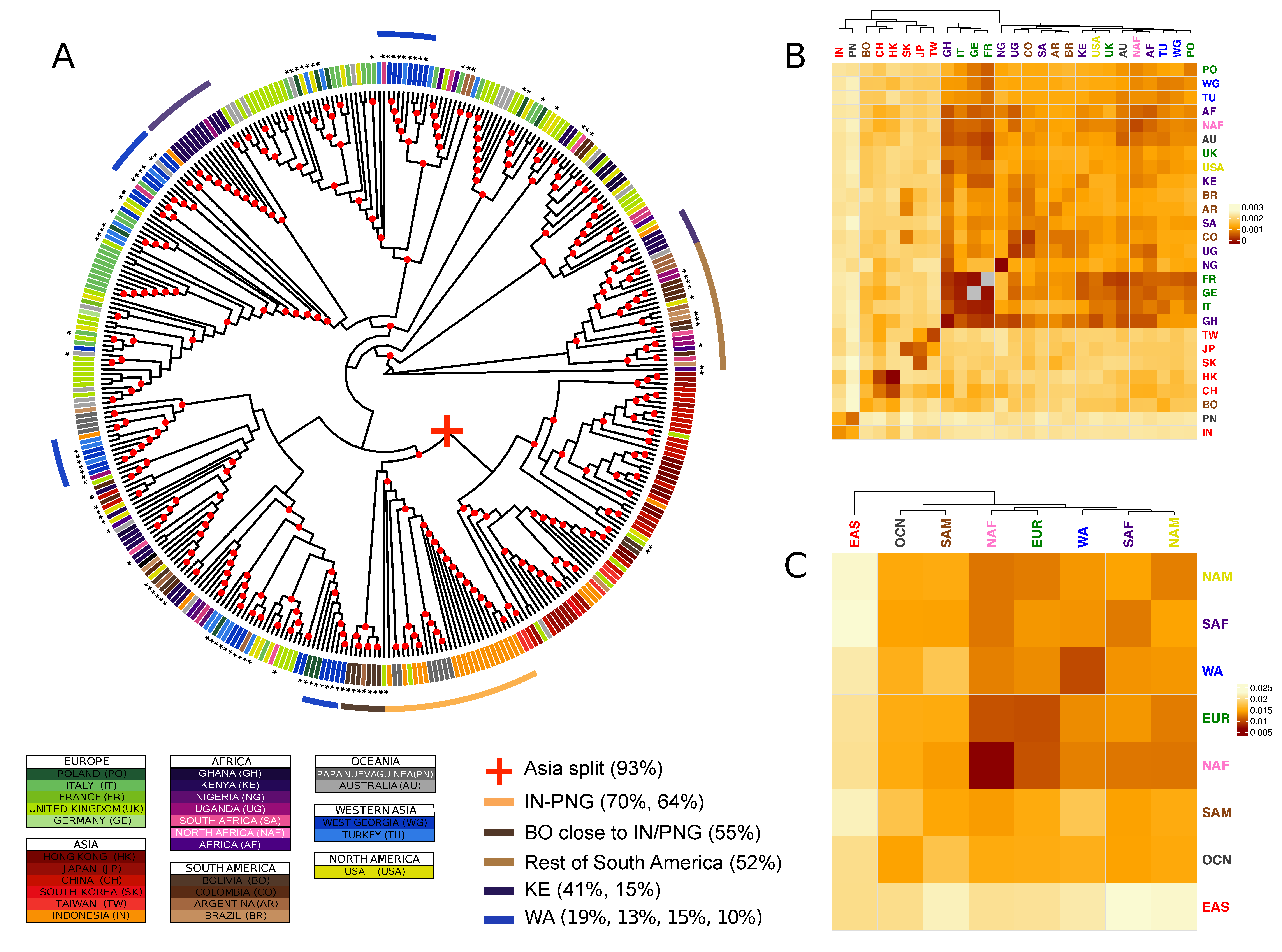
2.4. Population Structure: Phylogenetic Trees, Supertrees, and Population Pairwise Distances
2.5. Definition of Phylogeographic Group for Variant Identification
3. Results
3.1. Generation of EBV Sequences from Diverse Geographic Origins
3.2. Genetic Diversity
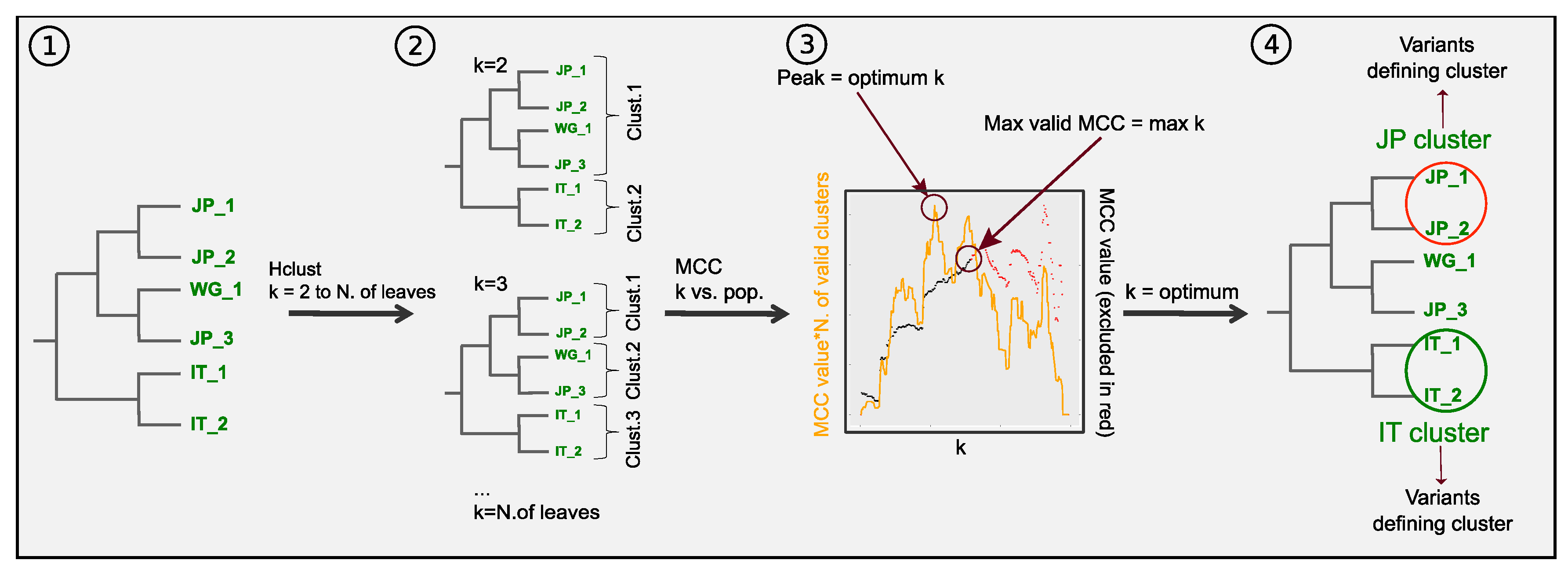
3.3. Combining Phylogenetic Information: Method Testing
3.4. Combining Phylogenetic Information: An Analytical Approach to Define Population Clusters
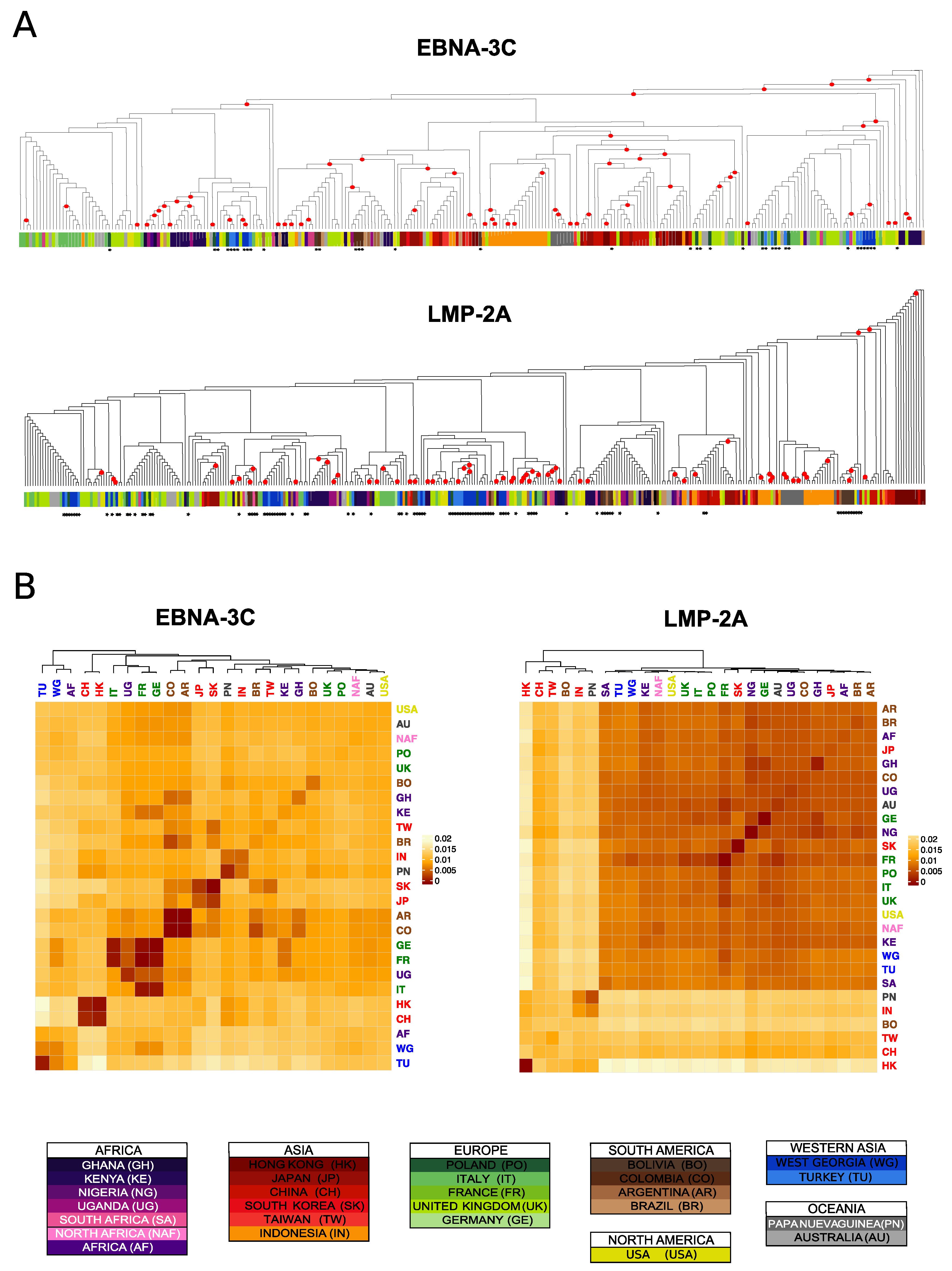
3.5. Differences and Commonalities in Individual Genes Phylogenies
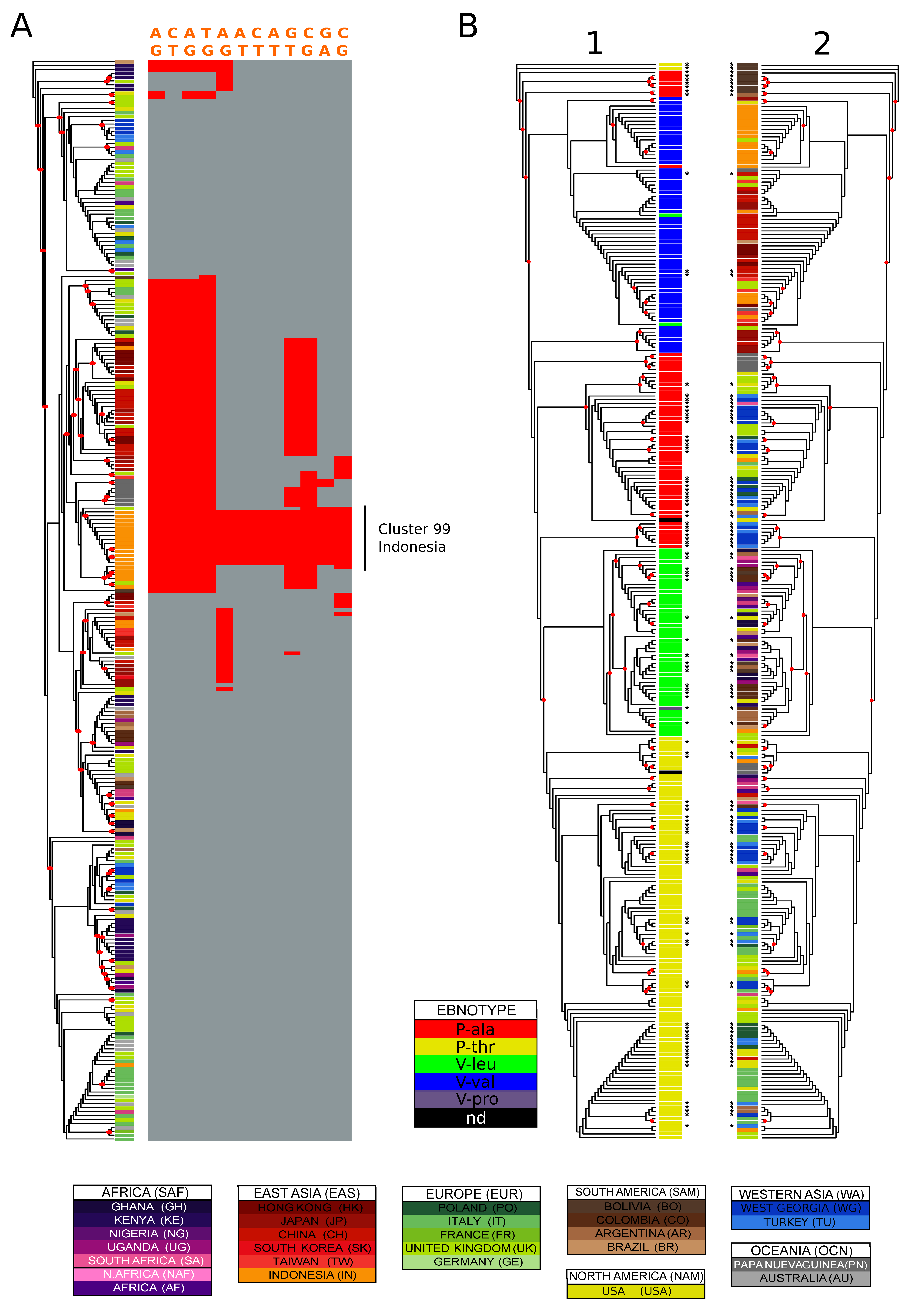
3.6. Identification of Geographically Informative Clusters and Genetic Variants
3.7. Classifying EBV Geographic Distribution
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chang, C.; Yu, K.; Mbulaiteye, S.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sültmann, H.; Moch, H.; Cooper, C.S.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P. Epstein-Barr virus and cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Jakimovski, D.; Ramanathan, M.; Weinstock-Guttman, B.; Zivadinov, R. The role of Epstein-Barr virus in multiple sclerosis: From molecular pathophysiology to in vivo imaging. Neural Regen. Res. 2019, 14, 373. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef]
- Giovannoni, G.; Ebers, G. Multiple sclerosis: The environment and causation. Curr. Opin. Neurol. 2007, 20, 261–268. [Google Scholar] [CrossRef]
- Cotsapas, C.; Mitrovic, M. Genome-wide association studies of multiple sclerosis. Clin. Transl. Immunol. 2018, 7, e1018. [Google Scholar] [CrossRef]
- Smith, P.R.; de Jesus, O.; Turner, D.; Hollyoake, M.; Karstegl, C.E.; Griffin, B.E.; Karran, L.; Wang, Y.; Hayward, S.D.; Farrell, P.J. Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J. Virol. 2000, 74, 3082–3092. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- White, R.E.; Rämer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bödör, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Investig. 2012, 122, 1487–1502. [Google Scholar] [CrossRef]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gärtner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous Lytic Replication and Epitheliotropism Define an Epstein-Barr Virus Strain Found in Carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheekollu, J.; Malecka, K.; Wiedmer, A.; Delecluse, H.-J.; Chiang, A.K.S.; Altieri, D.C.; Messick, T.E.; Lieberman, P.M. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr Virus episomal latency. Oncotarget 2017, 8, 7248–7264. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.-T.; Cui, Q.; Liu, W.-S.; Guo, Y.-M.; Feng, Q.-S.; Chen, L.-Z.; Xu, M.; Luo, B.; Li, D.-J.; Hu, L.-F.; et al. A single nucleotide polymorphism in the Epstein-Barr virus genome is strongly associated with a high risk of nasopharyngeal carcinoma. Chin. J. Cancer 2015, 34, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Yao, Y.; Chen, H.; Zhang, S.; Cao, S.-M.; Zhang, Z.; Luo, B.; Liu, Z.; Li, Z.; Xiang, T.; et al. Genome sequencing analysis identifies Epstein–Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 2019, 51, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, O.; Smith, P.R.; Spender, L.C.; Elgueta Karstegl, C.; Niller, H.H.; Huang, D.; Farrell, P.J. Updated Epstein-Barr virus (EBV) DNA sequence and analysis of a promoter for the BART (CST, BARF0) RNAs of EBV. J. Gen. Virol. 2003, 84, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.-S.; Li, D.-J.; Liu, Q.-L.; Song, L.-B.; Li, M.-Z.; Zhang, R.-H.; Yu, X.-J.; Wang, H.-M.; Ernberg, I.; Zeng, Y.-X. Genomic sequence analysis of Epstein-Barr virus strain GD1 from a nasopharyngeal carcinoma patient. J. Virol. 2005, 79, 15323–15330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, A.; Addison, C.; Gatherer, D.; Davison, A.J.; McGeoch, D.J. The genome of Epstein–Barr virus type 2 strain AG876. Virology 2006, 350, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Fang, X.; Feng, Z.; Guo, Y.-M.; Peng, R.-J.; Liu, T.; Huang, Z.; Feng, Y.; Sun, X.; Xiong, Z.; et al. Direct sequencing and characterization of a clinical isolate of Epstein-Barr virus from nasopharyngeal carcinoma tissue by using next-generation sequencing technology. J. Virol. 2011, 85, 11291–11299. [Google Scholar] [CrossRef] [Green Version]
- Kwok, H.; Tong, A.H.Y.; Lin, C.H.; Lok, S.; Farrell, P.J.; Kwong, D.L.W.; Chiang, A.K.S. Genomic Sequencing and Comparative Analysis of Epstein-Barr Virus Genome Isolated from Primary Nasopharyngeal Carcinoma Biopsy. PLoS ONE 2012, 7, e36939. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wang, X.; Strong, M.J.; Concha, M.; Baddoo, M.; Xu, G.; Baribault, C.; Fewell, C.; Hulme, W.; Hedges, D.; et al. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J. Virol. 2013, 87, 1172–1182. [Google Scholar] [CrossRef] [Green Version]
- Lei, H.; Li, T.; Li, B.; Tsai, S.; Biggar, R.J.; Nkrumah, F.; Neequaye, J.; Gutierrez, M.; Epelman, S.; Mbulaiteye, S.M.; et al. Epstein-Barr virus from Burkitt Lymphoma biopsies from Africa and South America share novel LMP-1 promoter and gene variations. Sci. Rep. 2015, 5, 16706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome Diversity of Epstein-Barr Virus from Multiple Tumor Types and Normal Infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiara, M.; Manzari, C.; Lionetti, C.; Mechelli, R.; Anastasiadou, E.; Buscarinu, M.C.; Ristori, G.; Salvetti, M.; Picardi, E.; D’Erchia, A.M.; et al. Geographic Population Structure in Epstein-Barr Virus Revealed by Comparative Genomics. Genome Biol. Evol. 2016, 8, 3284. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, W.; Pan, Y.; Ji, J.; Lu, Z.; Ke, Y. Genome-wide analysis of Epstein-Barr virus (EBV) isolated from EBV-associated gastric carcinoma (EBVaGC). Oncotarget 2016, 7, 4903–4914. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xiong, H.; Yan, S.; Wu, N.; Lu, Z. Identification and Characterization of Epstein-Barr Virus Genomes in Lung Carcinoma Biopsy Samples by Next-Generation Sequencing Technology. Sci. Rep. 2016, 6, 26156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Chen, J.-N.; Qiu, X.-M.; Pan, Y.-H.; Zhang, Z.-G.; Shao, C.-K. Comparative analysis of 22 Epstein-Barr virus genomes from diseased and healthy individuals. J. Gen. Virol. 2017, 98, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Bridges, R.; Wegner, F.; Venturini, C.; Palser, A.; Middeldorp, J.M.; Cohen, J.I.; Lorenzetti, M.A.; Bassano, I.; White, R.E.; et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Santpere, G.; Darre, F.; Blanco, S.; Alcami, A.; Villoslada, P.; Mar Albà, M.; Navarro, A.; Mar Alba, M.; Navarro, A.; Mar Albà, M.; et al. Genome-Wide Analysis of Wild-Type Epstein-Barr Virus Genomes Derived from Healthy Individuals of the 1000 Genomes Project. Genome Biol. Evol. 2014, 6, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Wegner, F.; Lassalle, F.; Depledge, D.P.; Balloux, F.; Breuer, J. Coevolution of Sites under Immune Selection Shapes Epstein–Barr Virus Population Structure. Mol. Biol. Evol. 2019, 36, 2512–2521. [Google Scholar] [CrossRef]
- Zanella, L.; Riquelme, I.; Buchegger, K.; Abanto, M.; Ili, C.; Brebi, P. A reliable Epstein-Barr Virus classification based on phylogenomic and population analyses. Sci. Rep. 2019, 9, 9829. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Young, L.S.; Cadwallader, K.; Petti, L.; Kieff, E.; Rickinson, A.B. Distinction between Epstein-Barr virus type A (EBNA 2A) and type B (EBNA 2B) isolates extends to the EBNA 3 family of nuclear proteins. J. Virol. 1989, 63, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, K.-M.; Li, C.-L.; Meng, G.; Han, A.-D.; Wu, X.-L. New BZLF1 sequence variations in EBV-associated undifferentiated nasopharyngeal carcinoma in southern China. Arch. Virol. 2008, 153, 1949–1953. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Xie, Z.; Lu, G.; Yang, S.; Shen, K. Characterization of variants in the promoter of BZLF1 gene of EBV in nonmalignant EBV-associated diseases in Chinese children. Virol. J. 2010, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.-Z.; Wang, Y.; Jia, Y.-P.; Luo, B. Polymorphisms of Epstein-Barr virus BHRF1 gene, a homologue of bcl-2. Chin. J. Cancer 2010, 29, 1000–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Tang, X.; Jia, Y.; Wang, Y.; Chao, Y.; Zhao, C. Sequence variation of Epstein-Barr virus (EBV) BZLF1 gene in EBV-associated gastric carcinomas and nasopharyngeal carcinomas in Northern China. Microbes Infect. 2011, 13, 776–782. [Google Scholar] [CrossRef]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.Z.T.; Niemann, J.; Iversen, K.H.; Fotakis, A.K.; Gopalakrishnan, S.; Vågene, Å.J.; Pedersen, M.W.; Sinding, M.-H.S.; Ellegaard, M.R.; Allentoft, M.E.; et al. A 5700 year-old human genome and oral microbiome from chewed birch pitch. Nat. Commun. 2019, 10, 5520. [Google Scholar] [CrossRef] [Green Version]
- Nasidze, I.; Li, J.; Quinque, D.; Tang, K.; Stoneking, M. Global diversity in the human salivary microbiome. Genome Res. 2009, 19, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carøe, C.; Gopalakrishnan, S.; Vinner, L.; Mak, S.S.T.; Sinding, M.H.S.; Samaniego, J.A.; Wales, N.; Sicheritz-Pontén, T.; Gilbert, M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2018, 9, 410–419. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup, 1000 Genome Project Data Processing The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541-8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Gorman, B. mltools: Machine Learning Tools 2018. Available online: https://cran.r-project.org/package=mltools (accessed on 18 October 2020).
- Arrand, J.R.; Young, L.S.; Tugwood, J.D. Two families of sequences in the small RNA-encoding region of Epstein-Barr virus (EBV) correlate with EBV types A and B. J. Virol. 1989, 63, 983–986. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.C.; Lin, S.C.; De, B.K.; Chan, W.P.; Evatt, B.L.; Chan, W.C. Precision of genotyping of Epstein-Barr virus by polymerase chain reaction using three gene loci (EBNA-2, EBNA-3C, and EBER): Predominance of type A virus associated with Hodgkin’s disease. Blood 1993, 81, 3372–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S. The effects of sample size on population genomic analyses – implications for the tests of neutrality. BMC Genom. 2016, 17, 123. [Google Scholar] [CrossRef] [Green Version]
- McGeoch, D.J.; Gatherer, D. Lineage structures in the genome sequences of three Epstein–Barr virus strains. Virology 2007, 359, 1–5. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Bhatia, K.; Raj, A.; Guitierrez, M.I.; Judde, J.G.; Spangler, G.; Venkatesh, H.; Magrath, I.T. Variation in the sequence of Epstein Barr virus nuclear antigen 1 in normal peripheral blood lymphocytes and in Burkitt’s lymphomas. Oncogene 1996, 13, 177–181. [Google Scholar]
- Aitken, C.; Sengupta, S.K.; Aedes, C.; Moss, D.J.; Sculley, T.B. Heterogeneity within the Epstein-Barr virus nuclear antigen 2 gene in different strains of Epstein-Barr virus. J. Gen. Virol. 1994, 75, 95–100. [Google Scholar] [CrossRef]
- Yamamoto, N.; Takizawa, T.; Iwanaga, Y.; Shimizu, N.; Yamamoto, N. Malignant transformation of B lymphoma cell line BJAB by Epstein-Barr virus-encoded small RNAs. FEBS Lett. 2000, 484, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Laing, K.G.; Elia, A.; Jeffrey, I.; Matys, V.; Tilleray, V.J.; Souberbielle, B.; Clemens, M.J. In Vivo Effects of the Epstein–Barr Virus Small RNA EBER-1 on Protein Synthesis and Cell Growth Regulation. Virology 2002, 297, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repellin, C.E.; Tsimbouri, P.M.; Philbey, A.W.; Wilson, J.B. Lymphoid Hyperplasia and Lymphoma in Transgenic Mice Expressing the Small Non-Coding RNA, EBER1 of Epstein-Barr Virus. PLoS ONE 2010, 5, e9092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, X.; Chao, Y.; Jia, Y.; Xing, X.; Luo, B. New variations of Epstein-Barr virus-encoded small RNA genes in nasopharyngeal carcinomas, gastric carcinomas, and healthy donors in northern China. J. Med. Virol. 2010, 82, 829–836. [Google Scholar] [CrossRef]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.S.; Colman, P.M. Structural Basis for Apoptosis Inhibition by Epstein-Barr Virus BHRF1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [Green Version]
- Lung, M.L.; Chang, R.S.; Jones, J.H. Genetic polymorphism of natural Epstein-Barr virus isolates from infectious mononucleosis patients and healthy carriers. J. Virol. 1988, 62, 3862–3866. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hamid, M.; Chen, J.-J.; Constantine, N.; Massoud, M.; Raab-traub, N. EBV strain variation: Geographical distribution and relation to disease state. Virology 1992, 190, 168–175. [Google Scholar] [CrossRef]
- Kwok, H.; Wu, C.W.; Palser, A.L.; Kellam, P.; Sham, P.C.; Kwong, D.L.W.; Chiang, A.K.S. Genomic Diversity of Epstein-Barr Virus Genomes Isolated from Primary Nasopharyngeal Carcinoma Biopsy Samples. J. Virol. 2014, 88, 10662–10672. [Google Scholar] [CrossRef] [Green Version]
- Walling, D.M.; Brown, A.L.; Etienne, W.; Keitel, W.A.; Ling, P.D. Multiple Epstein-Barr Virus Infections in Healthy Individuals. J. Virol. 2003, 77, 6546. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.; Palser, A.; Elgueta Karstegl, C.; Middeldorp, J.M.; Ramayanti, O.; Cohen, J.I.; Hildesheim, A.; Fellner, M.D.; Wiels, J.; White, R.E.; et al. Natural Variation of Epstein-Barr Virus Genes, Proteins, and Primary MicroRNA. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Midgley, R.S.; Blake, N.W.; Yao, Q.Y.; Croom-Carter, D.; Cheung, S.T.; Leung, S.F.; Chan, A.T.; Johnson, P.J.; Huang, D.; Rickinson, A.B.; et al. Novel intertypic recombinants of epstein-barr virus in the chinese population. J. Virol. 2000, 74, 1544–1548. [Google Scholar] [CrossRef] [Green Version]
- Lao, D.T.; Truong, K.P.; Huyen, T.A.L. Study of Epstein-Barr virus nuclear antigen (EBNA-1) variations: V-val type preferentially exists in biopsies of nasopharyngeal carcinoma from Vietnamese patients. Iran. J. Pediatr. Hematol. Oncol. 2018, 8, 75–86. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. Of Represented Strains | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | Country | Country_ID | Colour | BDLF4 | BGLF4 | BHRF1 | BRLF1 | BZLF1 | EBER-1 | EBER-2 | EBNA-1 | EBNA-2 * | EBNA-3A * | EBNA-3B * | EBNA-3C * | LMP-2 |
| EUROPE | Poland (10) | PO | 10 (10) | 10 (10) | 10 (10) | 10 (10) | 10 (10) | 9 (9) | 9 (9) | 10 (10) | 9 (9) | 10 (10) | 10 (10) | 8 (8) | 9 (9) | |
| Italy | IT | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | ||
| France | FR | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| UK | UK | 48 | 46 | 49 | 46 | 47 | 46 | 47 | 34 | 38 | 47 | 48 | 46 | 47 | ||
| Germany | GE | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| AFRICA | Ghana | GH | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 | 5 | 6 | |
| Kenya | KE | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 3 | 19 | 20 | 20 | 20 | 26 | ||
| Nigeria | NG | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 0 | 0 | 0 | 0 | 2 | ||
| Uganda | UG | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 3 | 5 | 5 | 5 | 6 | ||
| South Africa (4) | SA | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 2 (2) | 3 (3) | 3 (3) | 0 (0) | 4 (4) | ||
| North Africa ** | NAF | 7 | 7 | 7 | 7 | 7 | 6 | 6 | 6 | 5 | 6 | 6 | 6 | 6 | ||
| Africa ** | AF | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 6 | 5 | 5 | 5 | 4 | 7 | ||
| OCEANIA | Papa New Guinea | PN | 11 | 11 | 11 | 11 | 11 | 11 | 9 | 10 | 5 | 7 | 7 | 7 | 11 | |
| Australia | AU | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 0 | 19 | 19 | 19 | 19 | 20 | ||
| EAST ASIA | Hong Kong | HK | 11 | 11 | 11 | 11 | 11 | 11 | 11 | 5 | 11 | 11 | 11 | 11 | 11 | |
| Japan | JP | 10 | 11 | 12 | 10 | 10 | 10 | 12 | 10 | 8 | 11 | 11 | 11 | 12 | ||
| China (4) | CH | 24 (4) | 24 (4) | 24 (4) | 24 (4) | 24 (4) | 24 (4) | 23 (4) | 20 (4) | 22 (2) | 23 (3) | 23 (3) | 22 (2) | 24 (4) | ||
| South Korea | SK | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | ||
| Taiwan | TW | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 3 | 4 | 4 | 4 | 5 | ||
| Indonesia | IN | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 22 | 25 | 25 | 25 | 26 | ||
| SOUTH AMERICA | Bolivia (11) | BO | 11 (11) | 11 (11) | 10 (10) | 11 (11) | 11 (11) | 9 (9) | 9 (9) | 11 (11) | 7 (7) | 6 (6) | 5 (5) | 3 (3) | 10 (10) | |
| Colombia (12) | CO | 9 (9) | 12 (12) | 12 (12) | 10 (10) | 9 (9) | 9 (9) | 9 (9) | 11 (11) | 4 (4) | 4 (4) | 5 (5) | 4 (4) | 9 (9) | ||
| Argentina (6) | AR | 9 (6) | 9 (6) | 9 (6) | 8 (5) | 8 (5) | 8 (5) | 8 (5) | 9 (6) | 6 (3) | 6 (3) | 6 (3) | 4 (1) | 9 (6) | ||
| Brazil | BR | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | ||
| WESTERN ASIA | West Georgia (30) | WG | 29 (29) | 29 (29) | 28 (28) | 30 (30) | 30 (30) | 29 (29) | 29 (29) | 30 (30) | 16 (16) | 17 (17) | 18 (18) | 8 (8) | 29 (29) | |
| Turkey (18) | TU | 18 (18) | 18 (18) | 18 (18) | 18 (18) | 18 (18) | 16 (16) | 17 (17) | 18 (18) | 13 (13) | 14 (14) | 14 (14) | 9 (9) | 16 (16) | ||
| NORTH AMERICA | USA (8) | US | 23 (8) | 22 (7) | 22 (7) | 22 (7) | 23 (8) | 22 (7) | 22 (7) | 20 (8) | 21 (7) | 19 (4) | 19 (4) | 19 (4) | 23 (8) | |
| Genes | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lytic | Latent | ||||||||||||
| BDLF4 | BGLF4 | BHRF1 | BRLF1 | BZLF1 | EBER-1 | EBER-2 | EBNA-1 | EBNA-2 | EBNA-3A | EBNA-3B | EBNA-3C | LMP-2 | |
| Coding region without repeats (bp) | 678 | 1290 | 576 | 1818 | 738 | 167 | 173 | 1211 | 1340 | 2835 | 2641 | 2681 | 1494 |
| Callable region (bp) | 662 | 1189 | 576 | 1633 | 675 | 163 | 171 | 1187 | 1188 | 2257 | 1899 | 2392 | 1209 |
| Callable regions (%) | 98 | 92 | 100 | 90 | 91 | 98 | 99 | 98 | 89 | 80 | 72 | 89 | 81 |
| Polymorphic pos (S) | 49 | 61 | 67 | 117 | 66 | 8 | 27 | 143 | 205 | 307 | 229 | 263 | 184 |
| Genetic diversity (θW) | 0.0114 | 0.0079 | 0.0180 | 0.0111 | 0.0151 | 0.0076 | 0.0245 | 0.0193 | 0.0275 | 0.0214 | 0.0189 | 0.0175 | 0.0236 |
| New strains (N. of strains) | 99 | 101 | 99 | 99 | 99 | 92 | 93 | 102 | 63 | 64 | 65 | 39 | 95 |
| Published strains (N. of strains) | 262 | 261 | 265 | 260 | 261 | 259 | 259 | 184 | 234 | 262 | 263 | 260 | 260 |
| Final data set (Total N. Strains) | 361 | 362 | 364 | 359 | 360 | 351 | 352 | 286 | 279 (297) | 303 (326) | 305 (328) | 276 (299) | 355 |
| EBNA-1 | EBNA-2 | BHRF1 | EBER-2 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Res. 16 | Res. 487 | Insertion pos. 633/4 | Res. 79 | Res. 88 | Pos.44 | Pos. 46 | Pos. 57 | Pos. 61 | Pos. 93 | |||||||||||||||||
| Region | Country_ID | E | Q | A | L | P | T | V | CTC | CTT | No ins. | L | V | S | L | V | T | G | A | T | A | G | A | T | A | C |
| EUROPE | PO | 40 | 60 | 40 | 0 | 0 | 60 | 0 | 56 | 0 | 44 | 10 | 90 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 |
| IT | 4 | 96 | 4 | 0 | 0 | 96 | 0 | 67 | 0 | 33 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| FR | 0 | 100 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| UK | 38 | 62 | 26 | 6 | 0 | 50 | 18 | 58 | 0 | 42 | 2 | 98 | 0 | 90 | 10 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| GE | nd | nd | nd | nd | nd | nd | nd | 100 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| AFRICA | GH | 83 | 17 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 |
| KE | 67 | 33 | 0 | 67 | 0 | 33 | 0 | 63 | 0 | 38 | 4 | 96 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| NG | 100 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| UG | 83 | 17 | 0 | 83 | 0 | 17 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| SA | 75 | 25 | 25 | 50 | 0 | 25 | 0 | 50 | 0 | 50 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| NAF | 33 | 67 | 0 | 33 | 0 | 67 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| AF | 67 | 33 | 0 | 67 | 0 | 33 | 0 | 0 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| OCEANIA | PN | 50 | 50 | 50 | 0 | 0 | 20 | 20 | 0 | 0 | 100 | 82 | 18 | 0 | 100 | 0 | 78 | 22 | 78 | 22 | 78 | 22 | 78 | 22 | 78 | 22 |
| AU | nd | nd | nd | nd | nd | nd | nd | 60 | 0 | 40 | 0 | 100 | 0 | 95 | 5 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| EAST ASIA | HK | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 55 | 18 | 27 | 73 | 27 | 0 | 73 | 27 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 |
| JP | 50 | 50 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 0 | 8 | 92 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| CH | 10 | 90 | 0 | 10 | 0 | 15 | 75 | 9 | 0 | 91 | 17 | 83 | 0 | 25 | 75 | 78 | 22 | 78 | 22 | 78 | 22 | 78 | 22 | 78 | 22 | |
| SK | nd | nd | nd | nd | nd | nd | nd | 0 | 0 | 100 | 0 | 100 | 0 | 0 | 100 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| TW | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 100 | 0 | 100 | 0 | 20 | 80 | 80 | 20 | 80 | 20 | 80 | 20 | 80 | 20 | 80 | 20 | |
| IN | 11 | 89 | 7 | 4 | 0 | 15 | 74 | 46 | 0 | 54 | 56 | 44 | 0 | 78 | 22 | 85 | 15 | 85 | 15 | 85 | 15 | 85 | 15 | 85 | 15 | |
| SOUTH AMERICA | BO | 27 | 73 | 55 | 27 | 0 | 18 | 0 | 71 | 0 | 29 | 20 | 60 | 20 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 |
| CO | 91 | 9 | 0 | 82 | 9 | 9 | 0 | 25 | 0 | 75 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| AR | 67 | 33 | 22 | 56 | 0 | 22 | 0 | 17 | 0 | 83 | 11 | 89 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| BR | 67 | 33 | 0 | 67 | 0 | 17 | 17 | 33 | 0 | 67 | 33 | 67 | 0 | 100 | 0 | 83 | 17 | 83 | 17 | 83 | 17 | 83 | 17 | 83 | 17 | |
| WESTERN ASIA | WG | 40 | 60 | 53 | 0 | 0 | 47 | 0 | 38 | 0 | 63 | 29 | 71 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 |
| TU | 33 | 67 | 44 | 0 | 0 | 56 | 0 | 46 | 0 | 54 | 39 | 61 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | |
| NORTH AMERICA | US | 55 | 45 | 25 | 15 | 0 | 50 | 5 | 38 | 0 | 62 | 0 | 100 | 0 | 86 | 14 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 | 0 |
| TOTAL | 37.4 | 62.6 | 20.6 | 17.8 | 0.35 | 37.8 | 22.7 | 41.6 | 0.67 | 57.8 | 16.3 | 83.2 | 0.55 | 85.7 | 14.3 | 93.2 | 6.8 | 93.2 | 6.8 | 93.2 | 6.8 | 93.2 | 6.8 | 93.2 | 6.8 | |
| Sylthom_1 | nd | nd | + | - | - | - | - | nd | nd | nd | nd | nd | nd | nd | nd | + | - | + | - | + | - | nd | nd | nd | nd | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telford, M.; Hughes, D.A.; Juan, D.; Stoneking, M.; Navarro, A.; Santpere, G. Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches. Microorganisms 2020, 8, 1686. https://doi.org/10.3390/microorganisms8111686
Telford M, Hughes DA, Juan D, Stoneking M, Navarro A, Santpere G. Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches. Microorganisms. 2020; 8(11):1686. https://doi.org/10.3390/microorganisms8111686
Chicago/Turabian StyleTelford, Marco, David A. Hughes, David Juan, Mark Stoneking, Arcadi Navarro, and Gabriel Santpere. 2020. "Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches" Microorganisms 8, no. 11: 1686. https://doi.org/10.3390/microorganisms8111686
APA StyleTelford, M., Hughes, D. A., Juan, D., Stoneking, M., Navarro, A., & Santpere, G. (2020). Expanding the Geographic Characterisation of Epstein–Barr Virus Variation through Gene-Based Approaches. Microorganisms, 8(11), 1686. https://doi.org/10.3390/microorganisms8111686