
Corynebacterium glutamicum Regulation beyond Transcription: Organizing Principles and Reconstruction of an Extended Regulatory Network Incorporating Regulations Mediated by Small RNA and Protein–Protein Interactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
A Primer on Analyzing Regulatory Networks
2. Materials and Methods
2.1. Curation and Network Definition
2.2. Genome Annotation and Upstream Sequences
2.3. Regulatory Networks for Other Organisms
2.4. System-Level Components
2.5. Comparison of Nodes and Interactions of C. glutamicum with Other Bacterial Regulatory Networks
2.6. Global Structural Properties
2.7. System-Level Components
2.8. Regulog Analysis
3. Results and Discussion
3.1. The Regulatory Networks of C. glutamicum and Potential Applications
3.2. Global Networks of C. glutamicum Are Quite Different from other Bacterial Networks in Terms of Their Structural Properties
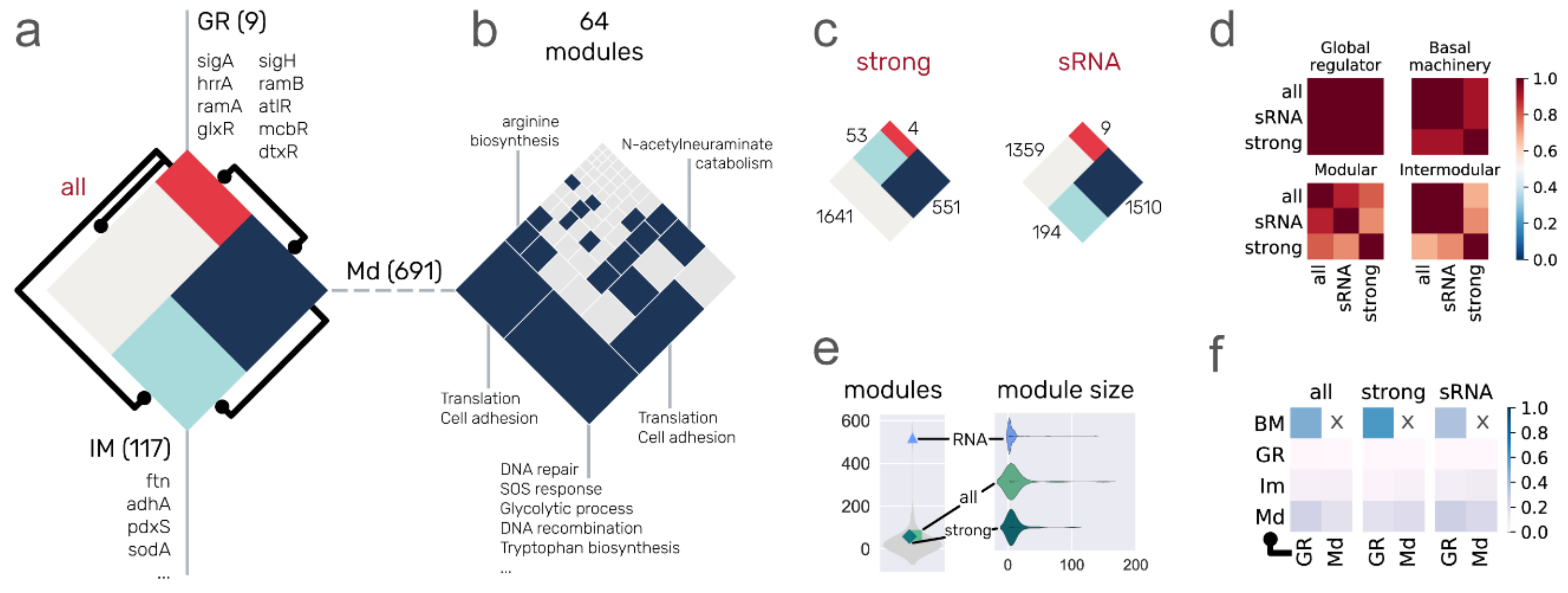
3.3. System-Level Components of the C. glutamicum Regulatory Networks
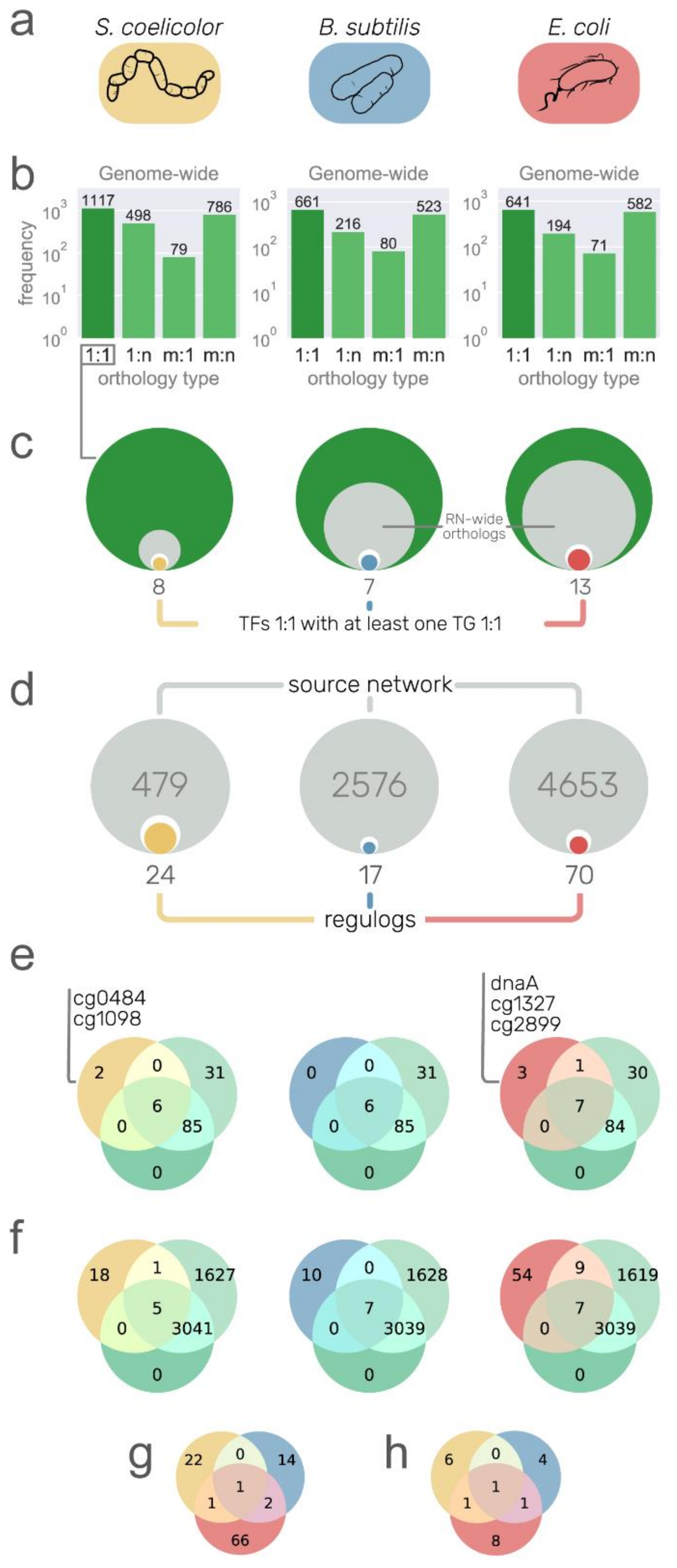
3.4. Recovering Conserved Interactions from Other Model Organisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escorcia-Rodríguez, J.M.; Tauch, A.; Freyre-González, J.A. Abasy Atlas v2.2: The most comprehensive and up-to-date inventory of meta-curated, historical, bacterial regulatory networks, their completeness and system-level characterization. Comput. Struct. Biotechnol. J. 2020, 18, 1228–1237. [Google Scholar] [CrossRef]
- Donovan, C.; Schauss, A.; Krämer, R.; Bramkamp, M. Chromosome Segregation Impacts on Cell Growth and Division Site Selection in Corynebacterium glutamicum. PLoS ONE 2013, 8, e55078. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, K.; Inui, M. Global Transcriptional Regulators Involved in Carbon, Nitrogen, Phosphorus, and Sulfur Metabolisms in Corynebacterium glutamicum. In Corynebacterium Glutamicum: Biology and Biotechnology; Inui, M., Toyoda, K., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 113–147. [Google Scholar] [CrossRef]
- Brinkrolf, K.; Sandbote, J.; Pühler, A.; Tauch, A. The transcriptional regulatory repertoire of Corynebacterium glutamicum: Reconstruction of the network controlling pathways involved in lysine and glutamate production. J. Biotechnol. 2010, 149, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Pátek, M.; Dostálová, H.; Nešvera, J. Sigma Factors of RNA Polymerase in Corynebacterium glutamicum. In Corynebacterium Glutamicum: Biology and Biotechnology; Inui, M., Toyoda, K., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 89–111. [Google Scholar] [CrossRef]
- Campos, A.I.; Freyre-González, J.A. Evolutionary constraints on the complexity of genetic regulatory networks allow predictions of the total number of genetic interactions. Sci. Rep. 2019, 9, 3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; DREAM5 Consortium; Kellis, M.; Collins, J.J.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.J.; Röttger, R.; Schmidt, H.H.H.W.; Baumbach, J. E. coli gene regulatory networks are inconsistent with gene expression data. Nucleic Acids Res. 2019, 47, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.; McCue, L.A.; Stormo, G.D. Making connections between novel transcription factors and their DNA motifs. Genome Res. 2005, 15, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodionov, D.A. Comparative Genomic Reconstruction of Transcriptional Regulatory Networks in Bacteria. Chem. Rev. 2007, 107, 3467–3497. [Google Scholar] [CrossRef]
- Alkema, W.; Lenhard, B.; Wasserman, W.W. Regulog Analysis: Detection of Conserved Regulatory Networks Across Bacteria: Application to Staphylococcus aureus. Genome Res. 2004, 14, 1362–1373. [Google Scholar] [CrossRef]
- Kılıç, S.; Erill, I. Assessment of transfer methods for comparative genomics of regulatory networks in bacteria. BMC Bioinform. 2016, 17 (Suppl. S8), 277. [Google Scholar] [CrossRef] [Green Version]
- Freyre-González, J.A.; Tauch, A. Functional architecture and global properties of the Corynebacterium glutamicum regulatory network: Novel insights from a dataset with a high genomic coverage. J. Biotechnol. 2017, 257, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzan, M.; Rehani, R.; Margalit, H. Integration of Bacterial Small RNAs in Regulatory Networks. Annu. Rev. Biophys. 2017, 46, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Parise, M.T.D.; Parise, D.; Kato, R.B.; Pauling, J.K.; Tauch, A.; Azevedo, V.A.D.C.; Baumbach, J. CoryneRegNet 7, the reference database and analysis platform for corynebacterial gene regulatory networks. Sci. Data 2020, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- De Witt, J.; Oetermann, S.; Parise, M.; Parise, D.; Baumbach, J.; Steinbüchel, A. Global Regulator of Rubber Degradation in Gordonia polyisoprenivorans VH2: Identification and Involvement in the Regulation Network. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J. The Structure and Function of Complex Networks. SIAM Rev. 2003, 45, 167–256. [Google Scholar] [CrossRef] [Green Version]
- Dorogovtsev, S.N.; Mendes, J.F.F. The shortest path to complex networks. arXiv 2004, arXiv:cond-mat/0404593. [Google Scholar]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Mason, O.; Verwoerd, M. Graph theory and networks in Biology. IET Syst. Biol. 2007, 1, 89–119. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.D.F.; Rodrigues, F.A.; Travieso, G.; Boas, P.R.V. Characterization of complex networks: A survey of measurements. Adv. Phys. 2007, 56, 167–242. [Google Scholar] [CrossRef] [Green Version]
- Lima-Mendez, G.; Van Helden, J. The powerful law of the power law and other myths in network biology. Mol. BioSyst. 2009, 5, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Freyre-González, J.A.; Alonso-Pavón, J.A.; Treviño-Quintanilla, L.G.; Collado-Vides, J. Functional architecture of Escherichia coli: New insights provided by a natural decomposition approach. Genome Biol. 2008, 9, R154. [Google Scholar] [CrossRef] [PubMed]
- Freyre-González, J.A.; Treviño-Quintanilla, L.G.; Valtierra-Gutiérrez, I.A.; Gutiérrez-Ríos, R.M.; Alonso-Pavón, J.A. Prokaryotic regulatory systems biology: Common principles governing the functional architectures of Bacillus subtilis and Escherichia coli unveiled by the natural decomposition approach. J. Biotechnol. 2012, 161, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mentz, A.; Neshat, A.; Pfeifer-Sancar, K.; Pühler, A.; Rückert, C.; Kalinowski, J. Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genom. 2013, 14, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coordinators, N.R.; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.T.; Contreras-Moreira, B.; Castro-Mondragon, J.A.; Santana-Garcia, W.; Ossio, R.; Robles-Espinoza, C.D.; Bahin, M.; Collombet, S.; Vincens, P.; Thieffry, D.; et al. RSAT 2018: Regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018, 46, W209–W214. [Google Scholar] [CrossRef] [Green Version]
- Baumbach, J.; Rahmann, S.; Tauch, A. Reliable transfer of transcriptional gene regulatory networks between taxonomically related organisms. BMC Syst. Biol. 2009, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, J.; Kohl, T.A.; Goesmann, A.; Kalinowski, J.; Baumbach, J. From Corynebacterium glutamicum to Mycobacterium tuberculosis—Towards transfers of gene regulatory networks and integrated data analyses with MycoRegNet. Nucleic Acids Res. 2009, 37, e97. [Google Scholar] [CrossRef]
- Altenhoff, A.; Levy, J.; Zarowiecki, M.; Tomiczek, B.; Vesztrocy, A.W.; Dalquen, D.A.; Müller, S.; Telford, M.J.; Glover, N.M.; Dylus, D.; et al. OMA standalone: Orthology inference among public and custom genomes and transcriptomes. Genome Res. 2019, 29, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Liu, X.; Brutlag, D.L.; Liu, J.S. BioProspector: Discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Biocomputing 2001, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Brutlag, D.L.; Liu, J.S. An algorithm for finding protein–DNA binding sites with applications to chromatin- immunoprecipitation microarray experiments. Nat. Biotechnol. 2002, 20, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastiri-Pancardo, G.; Mercado-Hernández, J.S.; Kim, J.; Jiménez, J.I.; Utrilla, J. A quantitative method for proteome reallocation using minimal regulatory interventions. Nat. Chem. Biol. 2020, 16, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, N.; Elowitz, M.B.; Alon, U. Negative Autoregulation Speeds the Response Times of Transcription Networks. J. Mol. Biol. 2002, 323, 785–793. [Google Scholar] [CrossRef]
- Ibarra-Arellano, M.A.; Campos-González, A.I.; Treviño-Quintanilla, L.G.; Tauch, A.; Freyre-González, J.A. Abasy Atlas: A comprehensive inventory of systems, global network properties and systems-level elements across bacteria. Database 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Orthologous Transcription Factors in Bacteria Have Different Functions and Regulate Different Genes. PLoS Comput. Biol. 2007, 3, e175. [Google Scholar] [CrossRef]
- Novichkov, P.S.; Kazakov, A.E.; Ravcheev, D.A.; Leyn, S.A.; Kovaleva, G.Y.; Sutormin, R.A.; Kazanov, M.D.; Riehl, W.; Arkin, A.P.; Dubchak, I.; et al. RegPrecise 3.0—A resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genom. 2013, 14, 745. [Google Scholar] [CrossRef]
- Baumbach, J. CoryneRegNet 4.0—A reference database for corynebacterial gene regulatory networks. BMC Bioinform. 2007, 8, 429. [Google Scholar] [CrossRef] [Green Version]
- Altenhoff, A.M.; Studer, R.A.; Robinson-Rechavi, M.; Dessimoz, C. Resolving the Ortholog Conjecture: Orthologs Tend to Be Weakly, but Significantly, More Similar in Function than Paralogs. PLoS Comput. Biol. 2012, 8, e1002514. [Google Scholar] [CrossRef] [Green Version]
- Bott, M.; Brocker, M. Two-component signal transduction in Corynebacterium glutamicum and other corynebacteria: On the way towards stimuli and targets. Appl. Microbiol. Biotechnol. 2012, 94, 1131–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, R.T.; Kriakov, J.; Garforth, S.J.; Baughn, A.D.; Jacobs, W.R., Jr. The Two-Component Regulatory System senX3-regX3 Regulates Phosphate-Dependent Gene Expression in Mycobacterium smegmatis. J. Bacteriol. 2007, 189, 5495–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parish, T.; Smith, D.A.; Roberts, G.; Betts, J.; Stoker, N.G. The senX3–regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 2003, 149, 1423–1435. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-García, A.; Sola-Landa, A.; Apel, K.; Santos-Beneit, F.; Martín, J.F. Phosphate control over nitrogen metabolism in Streptomyces coelicolor: Direct and indirect negative control of glnR, glnA, glnII and amtB expression by the response regulator PhoP. Nucleic Acids Res. 2009, 37, 3230–3242. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.; Küspert, M.; Ansorge, D.; Krämer, R.; Burkovski, A. Dissection of Ammonium Uptake Systems in Corynebacterium glutamicum: Mechanism of Action and Energetics of AmtA and AmtB. J. Bacteriol. 2008, 190, 2611–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Li, H.; Pan, Y.; Wang, L.; Tan, H.; Liu, G. SCO3129, a TetR family regulator, is responsible for osmotic stress in Streptomyces coelicolor. Synth. Syst. Biotechnol. 2018, 3, 261–267. [Google Scholar] [CrossRef]
- Cuthbertson, L.; Nodwell, J.R. The TetR Family of Regulators. Microbiol. Mol. Biol. Rev. 2013, 77, 440–475. [Google Scholar] [CrossRef] [Green Version]
- Kohl, T.A.; Tauch, A. The GlxR regulon of the amino acid producer Corynebacterium glutamicum: Detection of the corynebacterial core regulon and integration into the transcriptional regulatory network model. J. Biotechnol. 2009, 143, 239–246. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escorcia-Rodríguez, J.M.; Tauch, A.; Freyre-González, J.A. Corynebacterium glutamicum Regulation beyond Transcription: Organizing Principles and Reconstruction of an Extended Regulatory Network Incorporating Regulations Mediated by Small RNA and Protein–Protein Interactions. Microorganisms 2021, 9, 1395. https://doi.org/10.3390/microorganisms9071395
Escorcia-Rodríguez JM, Tauch A, Freyre-González JA. Corynebacterium glutamicum Regulation beyond Transcription: Organizing Principles and Reconstruction of an Extended Regulatory Network Incorporating Regulations Mediated by Small RNA and Protein–Protein Interactions. Microorganisms. 2021; 9(7):1395. https://doi.org/10.3390/microorganisms9071395
Chicago/Turabian StyleEscorcia-Rodríguez, Juan M., Andreas Tauch, and Julio A. Freyre-González. 2021. "Corynebacterium glutamicum Regulation beyond Transcription: Organizing Principles and Reconstruction of an Extended Regulatory Network Incorporating Regulations Mediated by Small RNA and Protein–Protein Interactions" Microorganisms 9, no. 7: 1395. https://doi.org/10.3390/microorganisms9071395