
Exploring the Individual Bacterial Microbiota of Questing Ixodes ricinus Nymphs

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Tick Sampling and Processing
2.2. DNA Extraction
2.3. PCR Amplification and High-Throughput Sequencing
2.4. Sequencing Data Processing
2.5. Borreliella Amplicon Sequence Variants Analysis
3. Results
3.1. Collection of Questing Ticks
3.2. Sequencing of the Internal Bacterial Microbiota of Ticks
3.3. Taxonomic Diversity in Nymphs
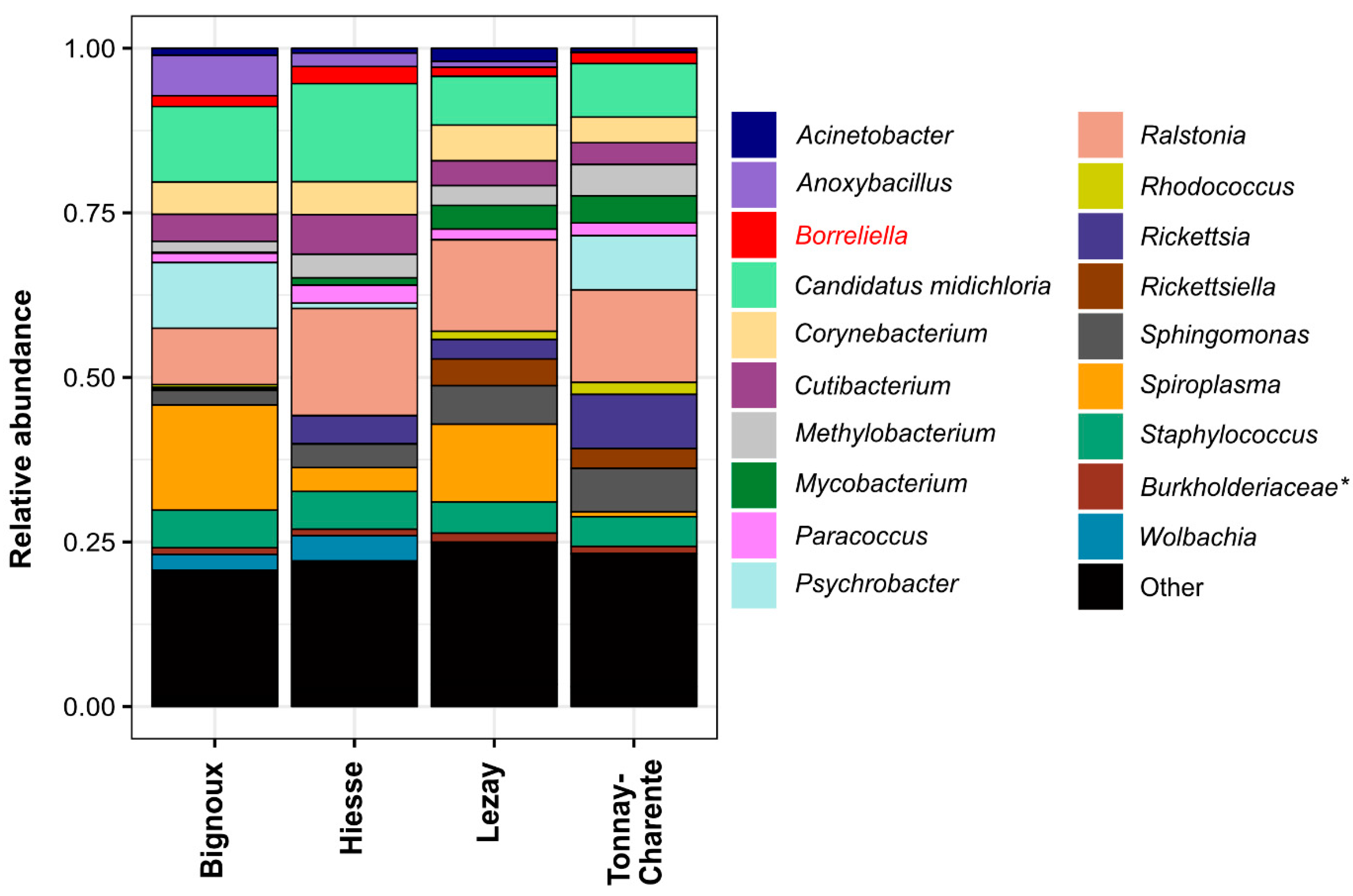
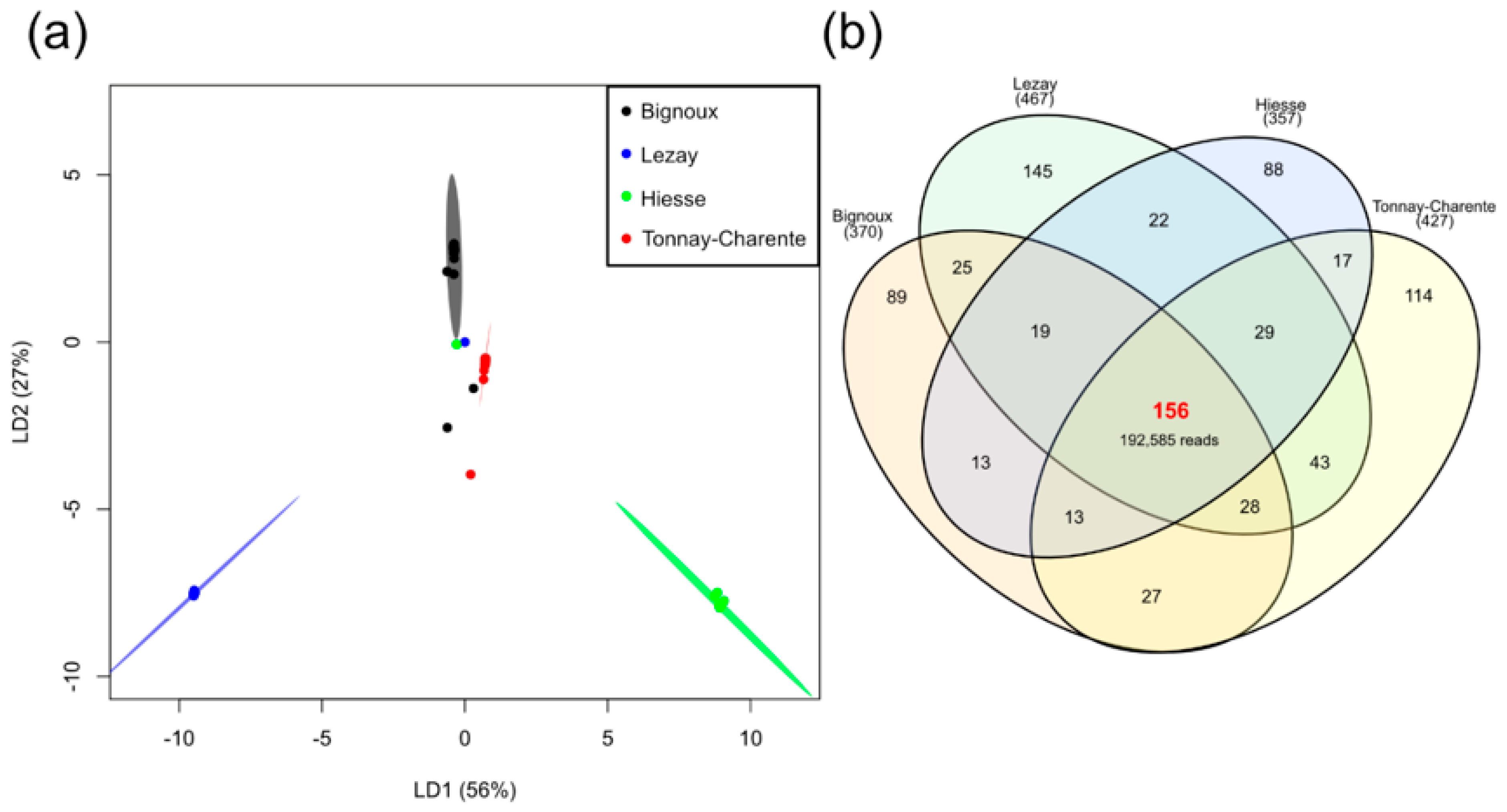
3.4. Site-Dependent Intra-Tick Diversity
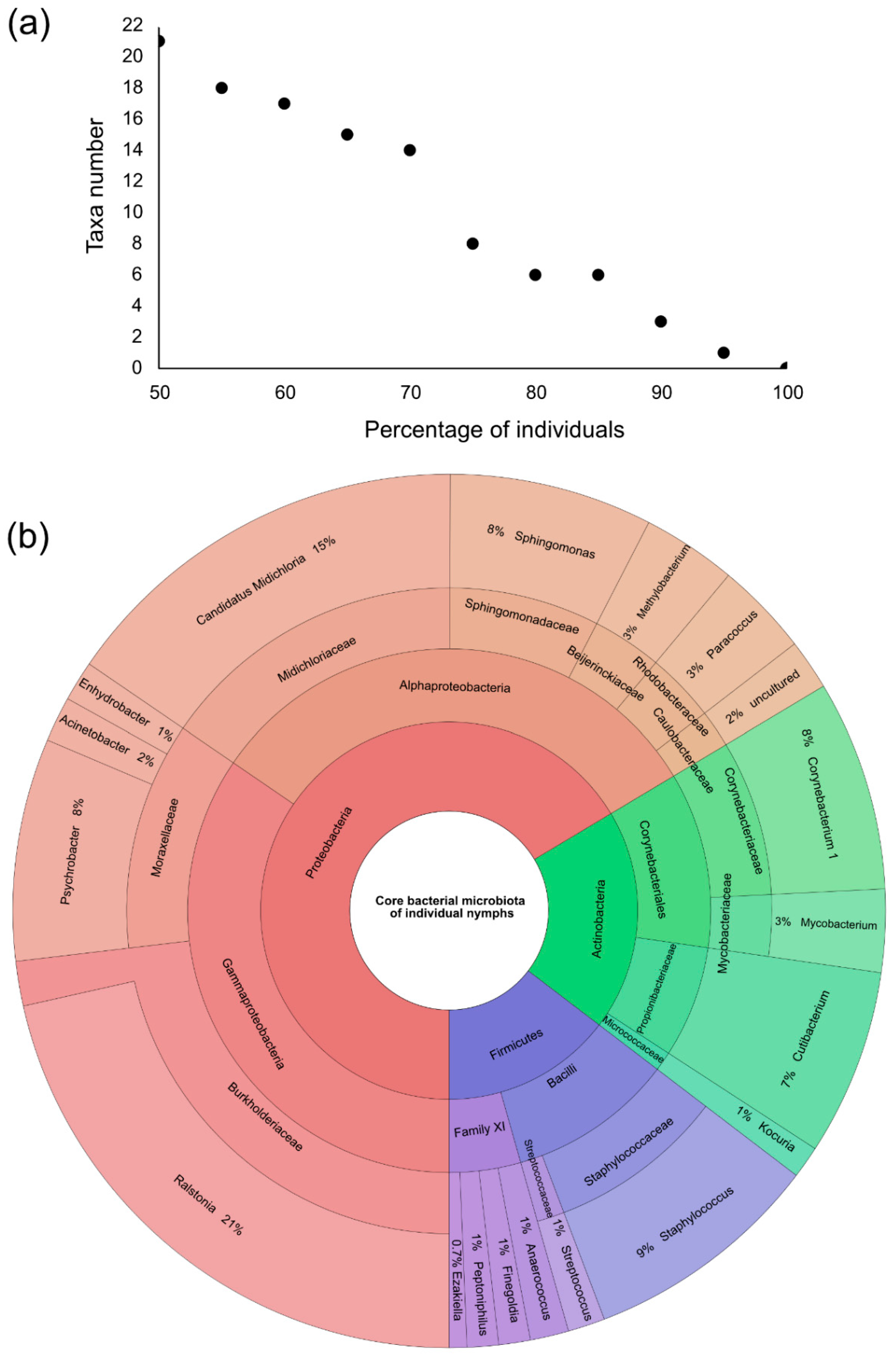
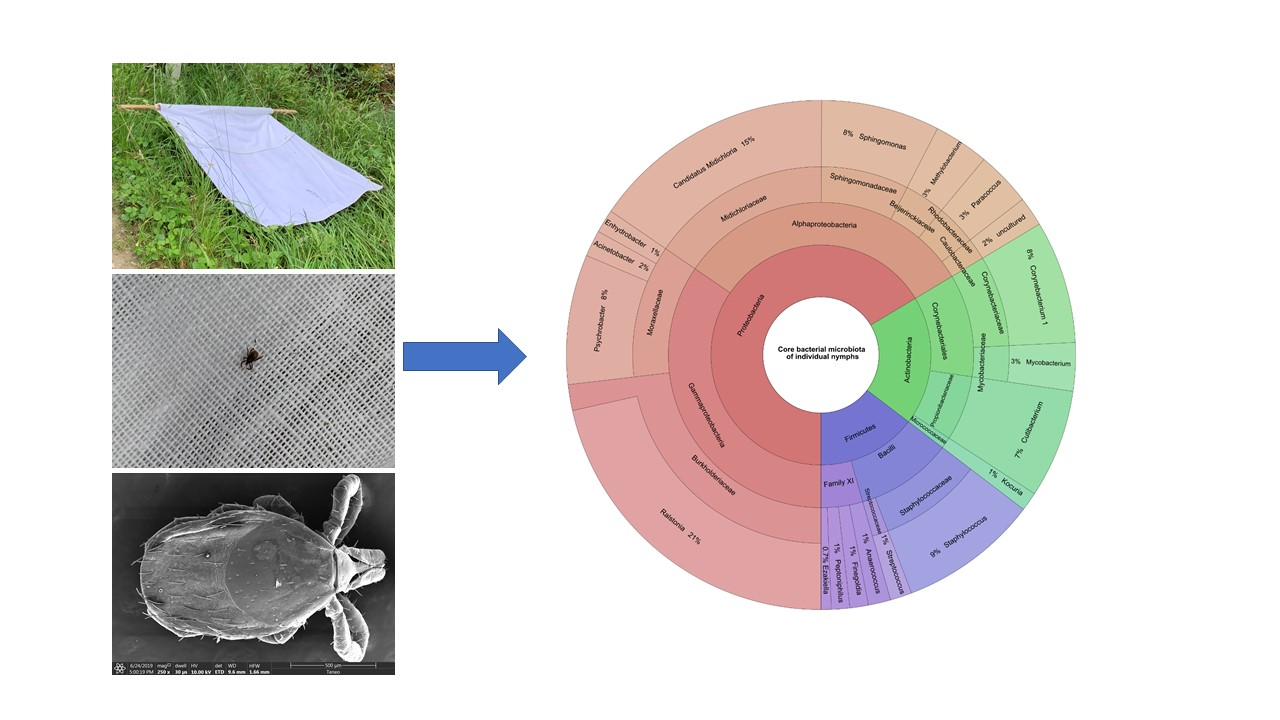
3.5. Individual Nymph Internal Core Microbiota
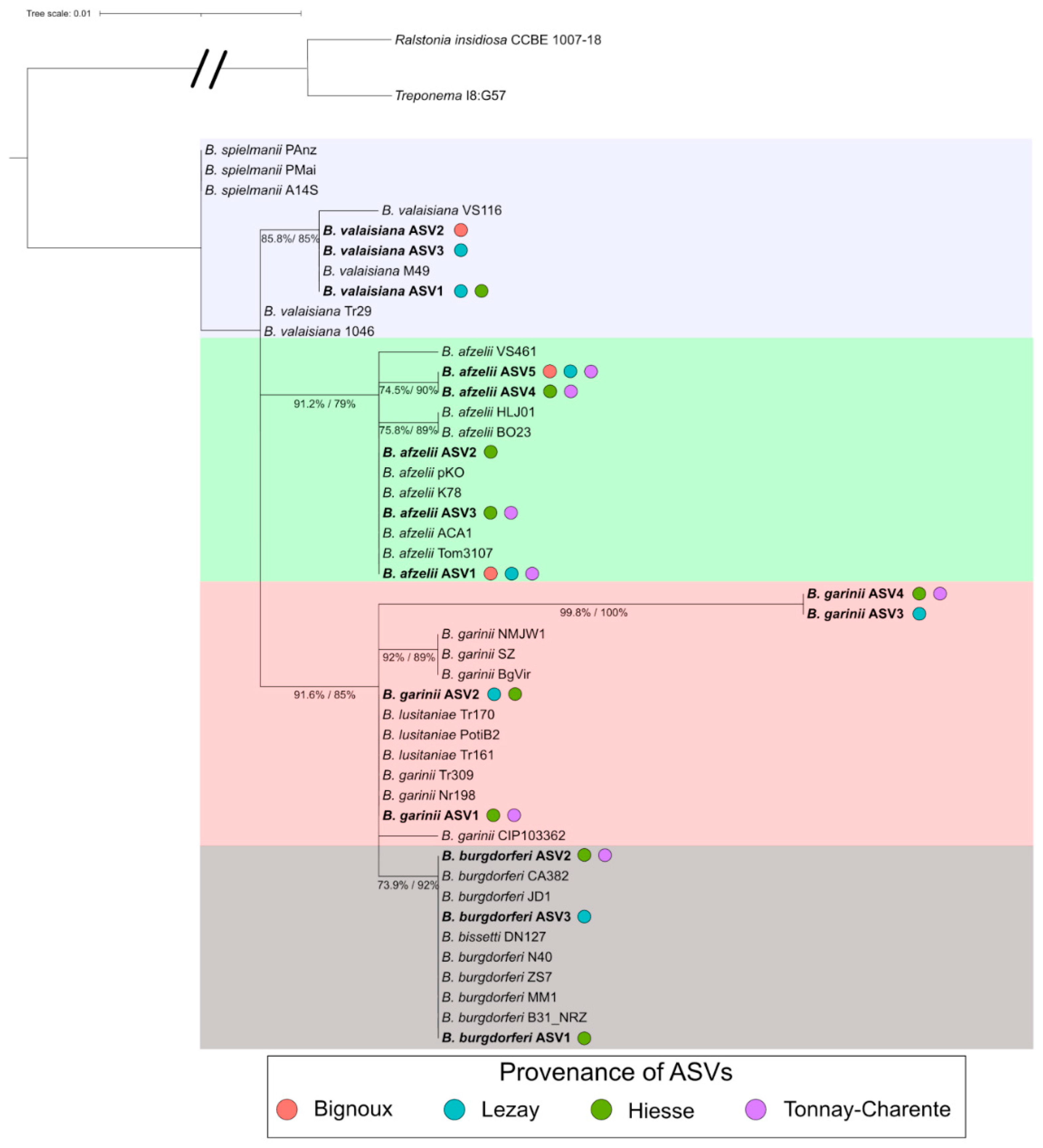
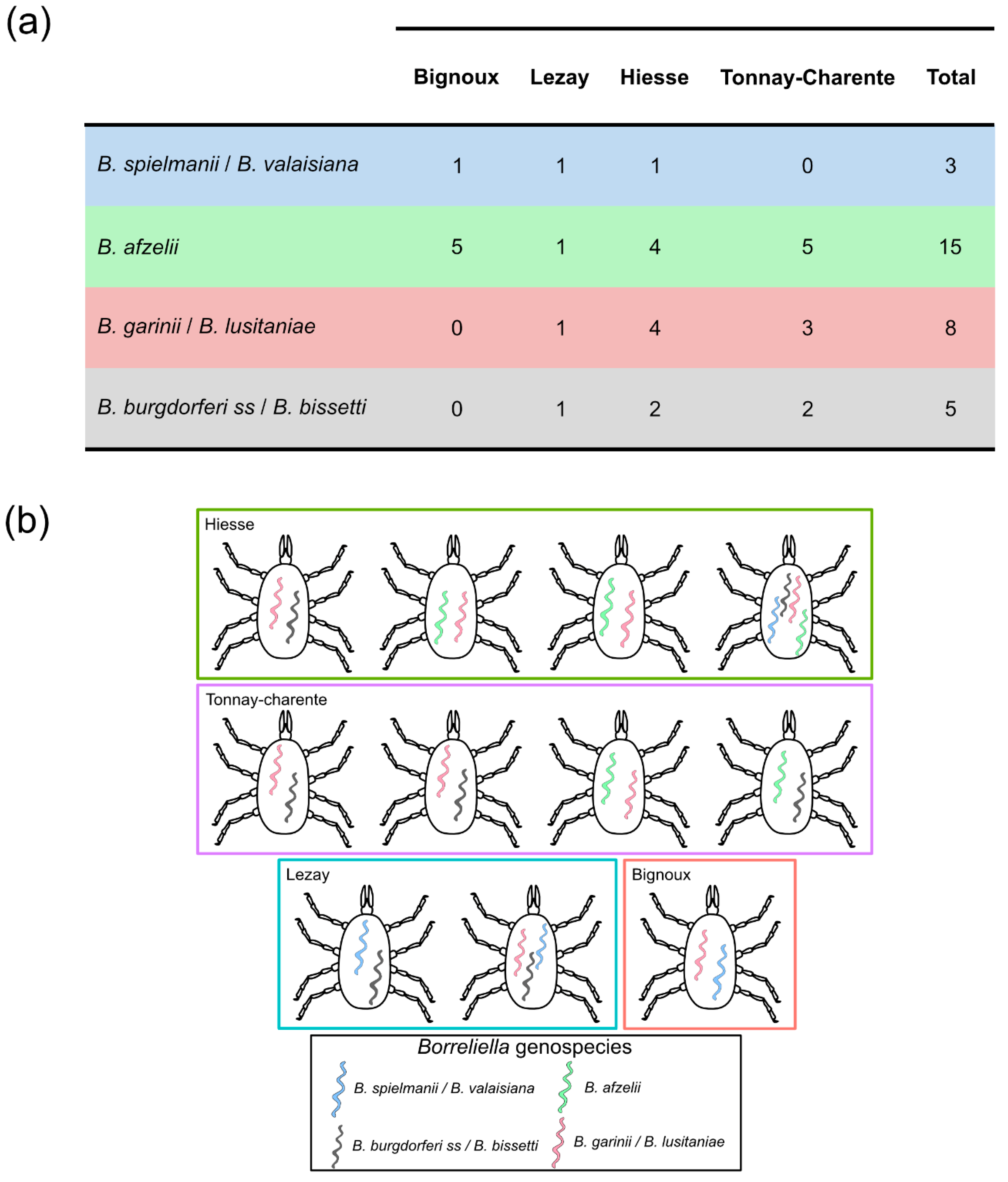
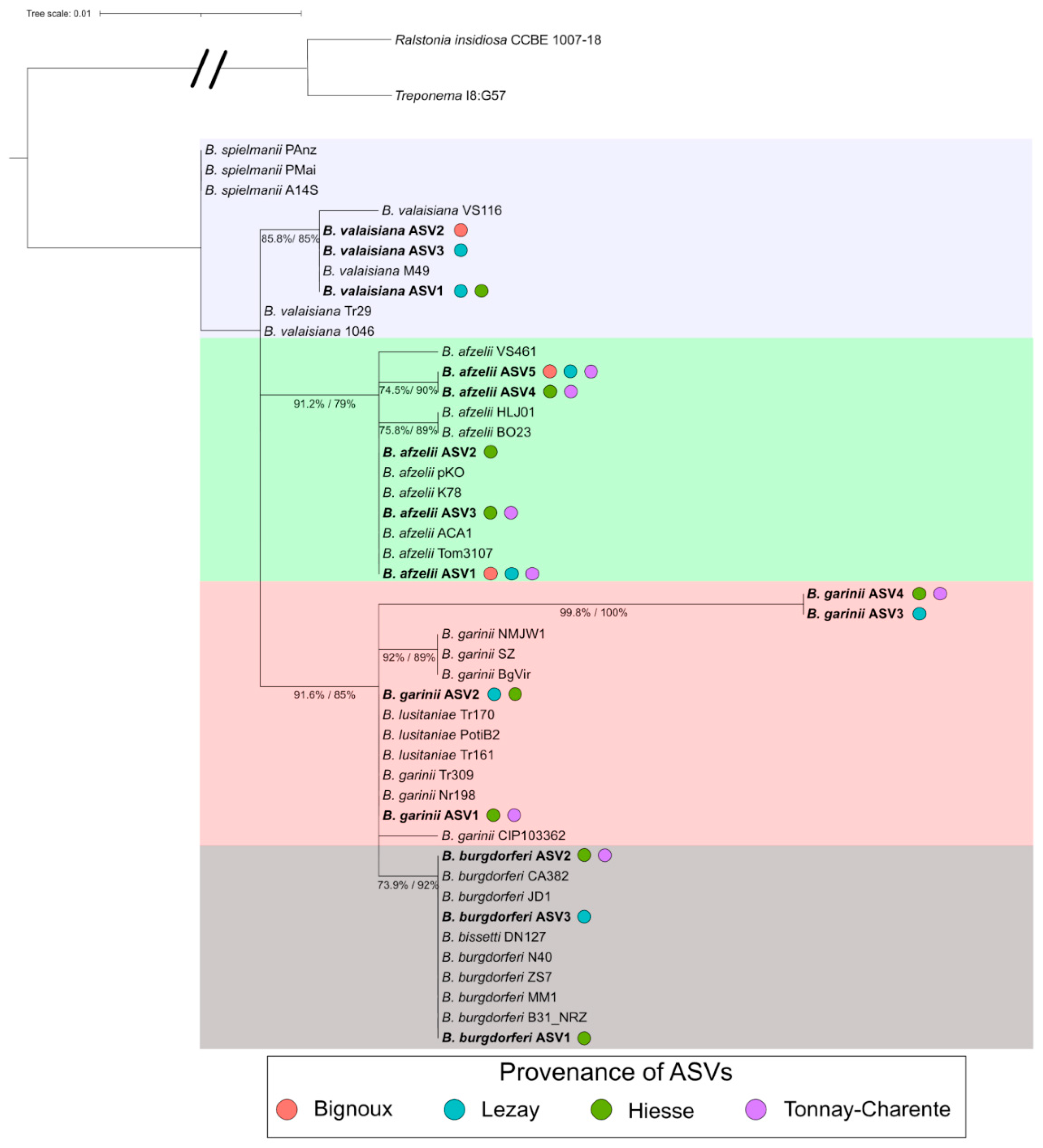
3.6. Borreliella Prevalence and Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, J.F.; Magnarelli, L.A. Biology of Ticks. Infect. Dis. Clin. N. Am. 2008, 22, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, N.; Boyer, P.; Talagrand-Reboul, E.; Hansmann, Y. Ticks and Tick-Borne Diseases. Med. Mal. Infect. 2019, 49, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, C.; Lynn, G.E.; Pedra, J.H.F.; Pal, U.; Narasimhan, S.; Fikrig, E. Interactions between Borrelia Burgdorferi and Ticks. Nat. Rev. Microbiol. 2020, 18, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Voordouw, M.J. Co-Feeding Transmission in Lyme Disease Pathogens. Parasitology 2015, 142, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent Insights into the Tick Microbiome Gained through Next-Generation Sequencing. Parasites Vectors 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, R.; De León, A.A.P.; Dowd, S.E.; Guerrero, F.D.; Bendele, K.G.; Scoles, G.A. Assessment of Bacterial Diversity in the Cattle Tick Rhipicephalus (Boophilus) Microplus through Tag-Encoded Pyrosequencing. BMC Microbiol. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Sánchez, S.; Estrada-Peña, A.; Cabezas-Cruz, A.; de la Fuente, J. Evolutionary Insights into the Tick Hologenome. Trends Parasitol. 2019, 35, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Sánchez, S.; Fernández, A.M.; Habela, M.A.; Calero-Bernal, R.; de Mera, I.G.F.; de la Fuente, J. Microbial Community of Hyalomma Lusitanicum Is Dominated by Francisella-like Endosymbiont. Ticks Tick. Borne. Dis. 2021, 12, 101624. [Google Scholar] [CrossRef]
- Binetruy, F.; Buysse, M.; Lejarre, Q.; Barosi, R.; Villa, M.; Rahola, N.; Paupy, C.; Ayala, D.; Duron, O. Microbial Community Structure Reveals Instability of Nutritional Symbiosis during the Evolutionary Radiation of Amblyomma Ticks. Mol. Ecol. 2020, 29, 1016–1029. [Google Scholar] [CrossRef]
- Gomard, Y.; Flores, O.; Vittecoq, M.; Blanchon, T.; Toty, C.; Duron, O.; Mavingui, P.; Tortosa, P.; McCoy, K.D. Changes in Bacterial Diversity, Composition and Interactions During the Development of the Seabird Tick Ornithodoros Maritimus (Argasidae). Microb. Ecol. 2021, 81, 770–783. [Google Scholar] [CrossRef]
- Landesman, W.J.; Mulder, K.; Page Fredericks, L.; Allan, B.F. Cross-Kingdom Analysis of Nymphal-Stage Ixodes Scapularis Microbial Communities in Relation to Borrelia Burgdorferi Infection and Load. FEMS Microbiol. Ecol. 2019, 95, fiz167. [Google Scholar] [CrossRef] [PubMed]
- Perveen, N.; Muzaffar, S.B.; Vijayan, R.; Al-Deeb, M.A. Microbial Communities Associated with the Camel Tick, Hyalomma Dromedarii: 16S RRNA Gene-Based Analysis. Sci. Rep. 2020, 10, 17035. [Google Scholar] [CrossRef] [PubMed]
- van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; Andreadis, T.G.; Falco, R.C.; Ziegler, L.B.; Hathaway, N.; et al. Variation in the Microbiota of Ixodes Ticks with Regard to Geography, Species, and Sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Yu, G.; Huang, Z.; Zhang, Z. Microbiota Assessment across Different Developmental Stages of Dermacentor Silvarum (Acari: Ixodidae) Revealed Stage-Specific Signatures. Ticks Tick. Borne. Dis. 2020, 11, 101321. [Google Scholar] [CrossRef] [PubMed]
- Pollet, T.; Sprong, H.; Lejal, E.; Krawczyk, A.I.; Moutailler, S.; Cosson, J.F.; Vayssier-Taussat, M.; Estrada-Peña, A. The Scale Affects Our View on the Identification and Distribution of Microbial Communities in Ticks. Parasites Vectors 2020, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, S.; Fikrig, E. Tick Microbiome: The Force Within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The Tick Microbiome: Why Non-Pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Duron, O.; Binetruy, F.; Noël, V.; Cremaschi, J.; McCoy, K.D.; Arnathau, C.; Plantard, O.; Goolsby, J.; Pérez de León, A.A.; Heylen, D.J.A.; et al. Evolutionary Changes in Symbiont Community Structure in Ticks. Mol. Ecol. 2017, 26, 2905–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duron, O.; Morel, O.; Noël, V.; Buysse, M.; Binetruy, F.; Lancelot, R.; Loire, E.; Ménard, C.; Bouchez, O.; Vavre, F.; et al. Tick-Bacteria Mutualism Depends on B Vitamin Synthesis Pathways. Curr. Biol. 2018, 28, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like Endosymbiontis a Potential Vitamin Source for the Lone Star Tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Thapa, S.; Zhang, Y.; Allen, M.S. Effects of Temperature on Bacterial Microbiome Composition in Ixodes Scapularis Ticks. Microbiologyopen 2018, 8, e719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkerhoff, R.J.; Clark, C.; Ocasio, K.; Gauthier, D.T.; Hynes, W.L. Factors Affecting the Microbiome of Ixodes Scapularis and Amblyomma Americanum. PLoS ONE 2020, 15, e0232398. [Google Scholar] [CrossRef]
- Hawlena, H.; Rynkiewicz, E.; Toh, E.; Alfred, A.; Durden, L.A.; Hastriter, M.W.; Nelson, D.E.; Rong, R.; Munro, D.; Dong, Q.; et al. The Arthropod, but Not the Vertebrate Host or Its Environment, Dictates Bacterial Community Composition of Fleas and Ticks. ISME J. 2013, 7, 221–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolnik, C.P.; Prill, R.J.; Falco, R.C.; Daniels, T.J.; Kolokotronis, S.O. Microbiome Changes through Ontogeny of a Tick Pathogen Vector. Mol. Ecol. 2016, 25, 4963–4977. [Google Scholar] [CrossRef]
- Obregón, D.; Bard, E.; Abrial, D.; Estrada-Peña, A.; Cabezas-Cruz, A. Sex-Specific Linkages Between Taxonomic and Functional Profiles of Tick Gut Microbiomes. Front. Cell. Infect. Microbiol. 2019, 9, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aivelo, T.; Norberg, A.; Tschirren, B. Bacterial Microbiota Composition of Ixodes Ricinus Ticks: The Role of Environmental Variation, Tick Characteristics and Microbial Interactions. PeerJ 2019, 2019, e8217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Ansari, J.M.; Jefferson, K.K.; Cava, F.; Jacobs-Wagner, C.; et al. Pathogen-Mediated Manipulation of Arthropod Microbiota to Promote Infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.I.; Pollet, T. Update on the Intricate Tango between Tick Microbiomes and Tick-Borne Pathogens. Parasite Immunol. 2021, 43, e12813. [Google Scholar] [CrossRef]
- Narasimhan, S.; Nallakkandi, R.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; DePonte, K.; Fish, D.; Fikrig, E. Gut Microbiota of the Tick Vector Ixodes Scapularis Modulate Colonization of the Lyme Disease Spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.J.; Daffre, S.; Hovius, J.W.; et al. Modulation of the Tick Gut Milieu by a Secreted Tick Protein Favors Borrelia Burgdorferi Colonization. Nat. Commun. 2017, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.M.; Krause, P.J.; Davis, S.; Vannier, E.G.; Fitzpatrick, M.C.; Rollend, L.; Belperron, A.A.; States, S.L.; Stacey, A.; Bockenstedt, L.K.; et al. Borrelia Burgdorferi Promotes the Establishment of Babesia Microti in the Northeastern United States. PLoS ONE 2014, 9, e115494. [Google Scholar] [CrossRef]
- Moutailler, S.; Valiente Moro, C.; Vaumourin, E.; Michelet, L.; Tran, F.H.; Devillers, E.; Cosson, J.F.; Gasqui, P.; Van, V.T.; Mavingui, P.; et al. Co-Infection of Ticks: The Rule Rather Than the Exception. PLoS Negl. Trop. Dis. 2016, 10, e0004539. [Google Scholar] [CrossRef] [Green Version]
- Raileanu, C.; Moutailler, S.; Pavel, I.; Porea, D.; Mihalca, A.D.; Savuta, G.; Vayssier-Taussat, M. Borrelia Diversity and Co-Infection with Other Tick Borne Pathogens in Ticks. Front. Cell. Infect. Microbiol. 2017, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Cosson, J.F.; Michelet, L.; Chotte, J.; Le Naour, E.; Cote, M.; Devillers, E.; Poulle, M.L.; Huet, D.; Galan, M.; Geller, J.; et al. Genetic Characterization of the Human Relapsing Fever Spirochete Borrelia Miyamotoi in Vectors and Animal Reservoirs of Lyme Disease Spirochetes in France. Parasites Vectors 2014, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarz, R.; Tagliafierro, T.; Sameroff, S.; Cucura, D.M.; Oleynik, A.; Che, X.; Jain, K.; Lipkin, W.I. Microbiome Analysis of Ixodes Scapularis Ticks from New York and Connecticut. Ticks Tick. Borne. Dis. 2019, 10, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Jahfari, S.; Hofhuis, A.; Fonville, M.; van der Giessen, J.; van Pelt, W.; Sprong, H. Molecular Detection of Tick-Borne Pathogens in Humans with Tick Bites and Erythema Migrans, in the Netherlands. PLoS Negl. Trop. Dis. 2016, 10, e0005042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, N.; Bhanot, P. Babesia Microti-Borrelia Burgdorferi. Pathogens 2019, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Cabezas-Cruz, A.; Vayssier-Taussat, M.; Greub, G. Tick-Borne Pathogen Detection: What’s New? Microbes Infect. 2018, 20, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Cutler, S.J.; Vayssier-Taussat, M.; Estrada-Peña, A.; Potkonjak, A.; Mihalca, A.D.; Zeller, H. Tick-Borne Diseases and Co-Infection: Current Considerations. Ticks Tick. Borne. Dis. 2021, 12, 101607. [Google Scholar] [CrossRef]
- Sykes, R.A.; Makiello, P. An Estimate of Lyme Borreliosis Incidence InWestern Europe. J. Public Heal. 2017, 39, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention Lyme Disease Data and Surveillance. Available online: https://www.cdc.gov/lyme/datasurveillance/recent-surveillance-data.html (accessed on 12 May 2021).
- Eisen, R.J.; Eisen, L.; Beard, C.B. County-Scale Distribution of Ixodes Scapularis and Ixodes Pacificus (Acari: Ixodidae) in the Continental United States. J. Med. Entomol. 2016, 53, 349–386. [Google Scholar] [CrossRef] [Green Version]
- Steere, A.C.; Strle, F.; Wormser, G.P.; Hu, L.T.; Branda, J.A.; Hovius, J.W.; Li, X.; Mead, P.S. Lyme Borreliosis. Nat. Rev. Dis. Prim. 2016, 2, 16090. [Google Scholar] [CrossRef]
- Barbour, A.G.; Adeolu, M.; Gupta, R.S. Division of the Genus Borrelia into Two Genera (Corresponding to Lyme Disease and Relapsing Fever Groups) Reflects Their Genetic and Phenotypic Distinctiveness and Will Lead to a Better Understanding of These Two Groups of Microbes. Int. J. Syst. Evol. Microbiol. 2017, 67, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.S.; Respicio-Kingry, L.B.; Sloan, L.M.; Schriefer, M.E.; Replogle, A.J.; Bjork, J.; Liu, G.; Kingry, L.C.; Mead, P.S.; Neitzel, D.F.; et al. Borrelia Mayonii Sp. Nov., a Member of the Borrelia Burgdorferi Sensu Lato Complex, Detected in Patients and Ticks in the Upper Midwestern United States. Int. J. Syst. Evol. Microbiol. 2016, 66, 4878–4880. [Google Scholar] [CrossRef]
- Stanek, G.; Fingerle, V.; Hunfeld, K.-P.; Jaulhac, B.; Kaiser, R.; Krause, A.; Kristoferitsch, W.; O’Connell, S.; Ornstein, K.; Strle, F.; et al. Lyme Borreliosis: Clinical Case Definitions for Diagnosis and Management in Europe. Clin. Microbiol. Infect. 2011, 17, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Stanek, G.; Wormser, G.P.; Gray, J.; Strle, F. Lyme Borreliosis. Lancet 2012, 379, 461–473. [Google Scholar] [CrossRef]
- Strnad, M.; Hönig, V.; Ruužek, D.; Grubhoffer, L.; Rego, R.O. Europe-Wide Meta-Analysis of Borrelia Burgdorferi Sensu Lato Prevalence in Questing Ixodes Ricinus Ticks. Appl. Environ. Microbiol. 2017, 83, e00609-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Eid, C. Les Tiques: Identification, Biologie, Importance Médicale et Vétérinaire; Tec & Doc Lavoisier, Ed.; Lavoisier: Paris, France, 2007. [Google Scholar]
- Binetruy, F.; Dupraz, M.; Buysse, M.; Duron, O. Surface Sterilization Methods Impact Measures of Internal Microbial Diversity in Ticks. Parasites Vectors 2019, 12, 268. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and Re-Analysis of Domain-Specific 16S Primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in Bacterial Communities along the 2000 Km Salinity Gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Perrin, Y.; Bouchon, D.; Delafont, V.; Moulin, L.; Héchard, Y. Microbiome of Drinking Water: A Full-Scale Spatio-Temporal Study to Monitor Water Quality in the Paris Distribution System. Water Res. 2019, 149, 375–385. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; Clark, K.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015, 43, D6–D17. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. BioRxiv 2018, 1–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pebesma, E.; Bivand, R.S. Classes and Methods for Spatial Data in R. R News 2005, 5, 9–13. [Google Scholar]
- South, A. Rnaturalearth: World Map Data from Natural Earth. 2017. Available online: https://CRAN-R-project.org/package=rnaturalearth (accessed on 13 April 2021).
- Becker, R.A.; Wilks, A.R. Maps: Draw Geographical Maps. 2018. Available online: https://CRAN-R-project.org/package=maps (accessed on 13 April 2021).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Oksanen, A.J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D.; Minchin, P.R.; Hara, R.B.O.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package; 2020; Available online: https://Cran-R-project.org/package=vegan (accessed on 11 January 2021).
- Kindt, R.; Coe, R. Tree Diversity Analysis; A Manual and Software for Common Statistical Methods for Ecological and Biodiversity Studies; 2005; Available online: https://www.worldagroforestry.org/iutput/tree-diversity-analysis (accessed on 11 January 2021).
- Venables, W.; Ripley, B. Modern Applied Statistics with S., 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, J.C.; Helal, Z.H.; Risatti, G.; Hird, S.M. Ixodes Scapularis Microbiome Correlates with Life Stage, Not the Presence of Human Pathogens, in Ticks Submitted for Diagnostic Testing. PeerJ 2020, 8, e10424. [Google Scholar] [CrossRef]
- Erb-Downward, J.R.; Falkowski, N.R.; D’souza, J.C.; McCloskey, L.M.; McDonald, R.A.; Brown, C.A.; Shedden, K.; Dickson, R.P.; Freeman, C.M.; Stringer, K.A.; et al. Critical Relevance of Stochastic Effects on Low-Bacterialbiomass 16s Rrna Gene Analysis. MBio 2020, 11, e00258-20. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and Laboratory Contamination Can Critically Impact Sequence-Based Microbiome Analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Akl, T.; Bourgoin, G.; Souq, M.L.; Appolinaire, J.; Poirel, M.T.; Gibert, P.; Abi Rizk, G.; Garel, M.; Zenner, L. Detection of Tick-Borne Pathogens in Questing Ixodes Ricinus in the French Pyrenees and First Identification of Rickettsia Monacensis in France. Parasite 2019, 26, 20. [Google Scholar] [CrossRef] [Green Version]
- Hvidsten, D.; Frafjord, K.; Gray, J.S.; Henningsson, A.J.; Jenkins, A.; Kristiansen, B.E.; Lager, M.; Rognerud, B.; Slåtsve, A.M.; Stordal, F.; et al. The Distribution Limit of the Common Tick, Ixodes Ricinus, and Some Associated Pathogens in North-Western Europe. Ticks Tick. Borne. Dis. 2020, 11, 101388. [Google Scholar] [CrossRef] [PubMed]
- Kjellander, P.L.; Aronsson, M.; Bergvall, U.A.; Carrasco, J.L.; Christensson, M.; Lindgren, P.E.; Åkesson, M.; Kjellander, P. Validating a Common Tick Survey Method: Cloth-Dragging and Line Transects. Exp. Appl. Acarol. 2021, 83, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Rulison, E.L.; Kuczaj, I.; Pang, G.; Hickling, G.J.; Tsao, J.I.; Ginsberg, H.S. Flagging versus Dragging as Sampling Methods for Nymphal Ixodes Scapularis (Acari: Ixodidae). J. Vector Ecol. 2013, 38, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Blazejak, K.; Janecek, E.; Strube, C. A 10-Year Surveillance of Rickettsiales (Rickettsia Spp. and Anaplasma Phagocytophilum) in the City of Hanover, Germany, Reveals Rickettsia Spp. as Emerging Pathogens in Ticks. Parasites Vectors 2017, 10, 588. [Google Scholar] [CrossRef] [Green Version]
- Dobson, A.D.M.; Taylor, J.L.; Randolph, S.E. Tick (Ixodes Ricinus) Abundance and Seasonality at Recreational Sites in the UK: Hazards in Relation to Fine-Scale Habitat Types Revealed by Complementary Sampling Methods. Ticks Tick. Borne. Dis. 2011, 2, 67–74. [Google Scholar] [CrossRef]
- Matuschka, F.-R.; Fischer, P.; Heiler, M.; Blümcke, S.; Spielman, A. Stage-Associated Risk of Transmission of the Lyme Disease Spirochete by European Ixodes Ticks*. Parasitol. Res. 1992, 78, 695–698. [Google Scholar] [CrossRef]
- Grech-Angelini, S.; Stachurski, F.; Vayssier-Taussat, M.; Devillers, E.; Casabianca, F.; Lancelot, R.; Uilenberg, G.; Moutailler, S. Tick-Borne Pathogens in Ticks (Acari: Ixodidae) Collected from Various Domestic and Wild Hosts in Corsica (France), a Mediterranean Island Environment. Transbound. Emerg. Dis. 2020, 67, 745–757. [Google Scholar] [CrossRef]
- Lejal, E.; Estrada-Peña, A.; Marsot, M.; Cosson, J.F.; Rué, O.; Mariadassou, M.; Midoux, C.; Vayssier-Taussat, M.; Pollet, T. Taxon Appearance From Extraction and Amplification Steps Demonstrates the Value of Multiple Controls in Tick Microbiota Analysis. Front. Microbiol. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Plantard, O.; Bouju-Albert, A.; Malard, M.A.; Hermouet, A.; Capron, G.; Verheyden, H. Detection of Wolbachia in the Tick Ixodes Ricinus Is Due to the Presence of the Hymenoptera Endoparasitoid Ixodiphagus Hookeri. PLoS ONE 2012, 7, e30692. [Google Scholar] [CrossRef] [Green Version]
- Bohacsova, M.; Mediannikov, O.; Kazimirova, M.; Raoult, D.; Sekeyova, Z. Arsenophonus Nasoniae and Rickettsiae Infection of Ixodes Ricinus Due to Parasitic Wasp Ixodiphagus Hookeri. PLoS ONE 2016, 11, e0149950. [Google Scholar] [CrossRef] [Green Version]
- Karim, S.; Budachetri, K.; Mukherjee, N.; Williams, J.; Kausar, A.; Hassan, M.J.; Adamson, S.; Dowd, S.E.; Apanskevich, D.; Arijo, A.; et al. A Study of Ticks and Tick-Borne Livestock Pathogens in Pakistan. PLoS Negl. Trop. Dis. 2017, 11, e0005681. [Google Scholar] [CrossRef]
- Moreno, C.X.; Moy, F.; Daniels, T.J.; Godfrey, H.P.; Cabello, F.C. Molecular Analysis of Microbial Communities Identified in Different Developmental Stages of Ixodes Scapularis Ticks from Westchester and Dutchess Counties, New York. Environ. Microbiol. 2006, 8, 761–772. [Google Scholar] [CrossRef]
- Peeters, N.; Guidot, A.; Vailleau, F.; Valls, M. Ralstonia Solanacearum, a Widespread Bacterial Plant Pathogen in the Post-Genomic Era. Mol. Plant Pathol. 2013, 14, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Sassera, D.; Beninati, T.; Bandi, C.; Bouman, E.A.P.; Sacchi, L.; Fabbi, M.; Lo, N. Candidatus Midichloria Mitochondrii’, an Endosymbiont of the Ixodes Ricinus with a Unique Intramitochondrial Lifestyle. Int. J. Syst. Evol. Microbiol. 2006, 56, 2535–2540. [Google Scholar] [CrossRef]
- Lo, N.; Beninati, T.; Sassera, D.; Bouman, E.A.P.; Santagati, S.; Gern, L.; Sambri, V.; Masuzawa, T.; Gray, J.S.; Jaenson, T.G.T.; et al. Widespread Distribution and High Prevalence of an Alpha-Proteobacterial Symbiont in the Tick Ixodes Ricinus. Environ. Microbiol. 2006, 8, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Stavru, F.; Riemer, J.; Jex, A.; Sassera, D. When Bacteria Meet Mitochondria: The Strange Case of the Tick Symbiont Midichloria Mitochondrii†. Cell. Microbiol. 2020, 22, e13189. [Google Scholar] [CrossRef] [Green Version]
- Budachetri, K.; Kumar, D.; Crispell, G.; Beck, C.; Dasch, G.; Karim, S. The Tick Endosymbiont Candidatus Midichloria Mitochondrii and Selenoproteins Are Essential for the Growth of Rickettsia Parkeri in the Gulf Coast Tick Vector. Microbiome 2018, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Tufts, D.M.; Sameroff, S.; Tagliafierro, T.; Jain, K.; Oleynik, A.; VanAcker, M.C.; Diuk-Wasser, M.A.; Lipkin, W.I.; Tokarz, R. A Metagenomic Examination of the Pathobiome of the Invasive Tick Species, Haemaphysalis Longicornis, Collected from a New York City Borough, USA. Ticks Tick. Borne. Dis. 2020, 11, 101516. [Google Scholar] [CrossRef]
- Keskin, A.; Bursali, A.; Snow, D.E.; Dowd, S.E.; Tekin, S. Assessment of Bacterial Diversity in Hyalomma Aegyptium, H. Marginatum and H. Excavatum Ticks through Tag-Encoded Pyrosequencing. Exp. Appl. Acarol. 2017, 73, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Shames, S.R. Pathogenicity and Virulence of Legionella: Intracellular Replication and Host Response. Virulence 2021, 12, 1122–1144. [Google Scholar] [CrossRef]
- Casati, S.; Gioria-Martinoni, A.; Gaia, V. Commercial Potting Soils as an Alternative Infection Source of Legionella Pneumophila and Other Legionella Species in Switzerland. Clin. Microbiol. Infect. 2009, 15, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Remesar, S.; Díaz, P.; Portillo, A.; Santibáñez, S.; Prieto, A.; Díaz-Cao, J.M.; López, C.M.; Panadero, R.; Fernández, G.; Díez-Baños, P.; et al. Prevalence and Molecular Characterization of Rickettsia Spp. in Questing Ticks from North-Western Spain. Exp. Appl. Acarol. 2019, 79, 267–278. [Google Scholar] [CrossRef]
- Katargina, O.; Geller, J.; Ivanova, A.; Värv, K.; Tefanova, V.; Vene, S.; Lundkvist, Å.; Golovljova, I. Detection and Identification of Rickettsia Species in Ixodes Tick Populations from Estonia. Ticks Tick. Borne. Dis. 2015, 6, 689–694. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Cutler, S.; Potkonjak, A.; Vassier-Tussaut, M.; Bortel, W.; Zeller, H.; Fernández-Ruiz, N.; Mihalca, A.D. An Updated Meta-Analysis of the Distribution and Prevalence of Borrelia Burgdorferi s.l. in Ticks in Europe. Int. J. Health Geogr. 2018, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Peña, A.; Ortega, C.; Sánchez, N.; DeSimone, L.; Sudre, B.; Suk, J.E.; Semenza, J.C. Correlation of Borrelia Burgdorferi Sensu Lato Prevalence in Questing Ixodes Ricinus Ticks with Specific Abiotic Traits in the Western Palearctic. Appl. Environ. Microbiol. 2011, 77, 3838–3845. [Google Scholar] [CrossRef] [Green Version]
- Rauter, C.; Hartung, T. Prevalence of Borrelia Burgdorferi Sensu Lato Genospecies in Ixodes Ricinus Ticks in Europe: A Metaanalysis. Appl. Environ. Microbiol. 2005, 71, 7203–7216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vayssier-Taussat, M.; Kazimirova, M.; Hubalek, Z.; Hornok, S.; Farkas, R.; Cosson, J.F.; Bonnet, S.; Vourc’h, G.; Gasqui, P.; Mihalca, A.D.; et al. Emerging Horizons for Understanding Tick-Borne Pathogens Threat: Shifting from the “One Pathogen-One Disease” Vision to the Pathobiome Paradigm. Future Microbiol. 2015, 10, 2033–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, J.; Moldenhauer, A.; Hildebrandt, A.; Dorn, W. Are Birds Reservoir Hosts for Borrelia Afzelii? Ticks Tick. Borne. Dis. 2010, 1, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Gern, L. Borrelia Burgdorferi Sensu Lato, the Agent of Lyme Borreliosis: Life in the Wilds. Parasite 2008, 15, 244–247. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Number of Individuals | Average Reads per Tick | Shannon H Index (±SD 1) | Shannon E Index (±SD 1) | Observed ASVs |
|---|---|---|---|---|---|
| Bignoux | 45 | 1055 | 2.83 (±0.81) | 0.77 (±0.15) | 1079 |
| Lezay | 43 | 1167 | 3.25 (±0.71) | 0.76 (±0.13) | 1371 |
| Hiesse | 43 | 1043 | 2.90 (±0.78) | 0.73 (±0.14) | 1019 |
| Tonnay-Charente | 44 | 1394 | 3.21 (±0.80) | 0.75 (±0.15) | 1433 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alafaci, A.; Crépin, A.; Beaubert, S.; Berjeaud, J.-M.; Delafont, V.; Verdon, J. Exploring the Individual Bacterial Microbiota of Questing Ixodes ricinus Nymphs. Microorganisms 2021, 9, 1526. https://doi.org/10.3390/microorganisms9071526
Alafaci A, Crépin A, Beaubert S, Berjeaud J-M, Delafont V, Verdon J. Exploring the Individual Bacterial Microbiota of Questing Ixodes ricinus Nymphs. Microorganisms. 2021; 9(7):1526. https://doi.org/10.3390/microorganisms9071526
Chicago/Turabian StyleAlafaci, Aurélien, Alexandre Crépin, Sabine Beaubert, Jean-Marc Berjeaud, Vincent Delafont, and Julien Verdon. 2021. "Exploring the Individual Bacterial Microbiota of Questing Ixodes ricinus Nymphs" Microorganisms 9, no. 7: 1526. https://doi.org/10.3390/microorganisms9071526
APA StyleAlafaci, A., Crépin, A., Beaubert, S., Berjeaud, J.-M., Delafont, V., & Verdon, J. (2021). Exploring the Individual Bacterial Microbiota of Questing Ixodes ricinus Nymphs. Microorganisms, 9(7), 1526. https://doi.org/10.3390/microorganisms9071526