
Fecal Supernatant from Adult with Autism Spectrum Disorder Alters Digestive Functions, Intestinal Epithelial Barrier, and Enteric Nervous System

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Study Approval
2.2. Subjects, Feces Collection, and Preparation of Fecal Supernatant (FS)
2.3. Microbiota and Bacterial Metabolite Analysis
2.3.1. 16S rRNA Gene Sequencing
2.3.2. Data Processing
2.3.3. Bacterial Metabolite Analysis
2.4. Microbiota-Metabolite Association
2.5. Mice Model of Fecal Supernatant Administration
2.6. In Vivo Intestinal Motility Assay
2.7. Intestinal Permeability Assay
2.7.1. In Vivo Intestinal Permeability
2.7.2. Ex Vivo Intestinal Permeability
2.8. ENS Primary Cultures and Treatment
2.9. mRNA and Proteins Extraction
2.10. Quantitative Real-Time PCR (RT-qPCR)
2.11. Western Blot
2.12. Histological Analysis
2.13. Statistical Analysis
3. Results
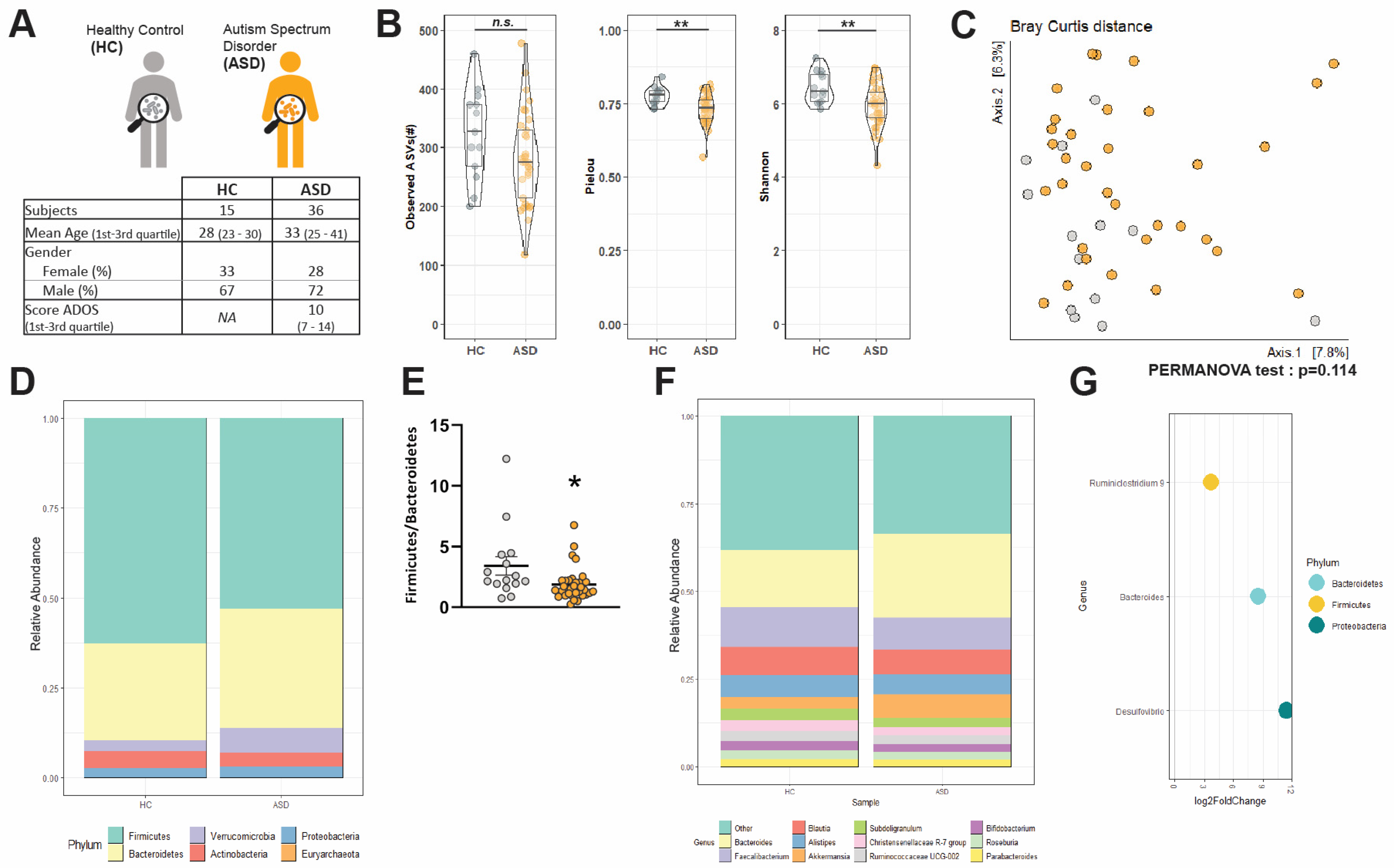
3.1. Socio-Demographic Data
3.2. Changes in α-Diversityand Bacterial Abundance in the Fecal Microbiota of Adult ASD Patients
3.3. Short Chain Fatty Acids and Bile Acids Are Unchanged in FS–ASD as Compared to FS–HC
3.4. Transfer of FS–ASD in Mice Induces Changes in Colonic Permeability
3.5. Transfer of FS–ASD in Mice Modulates Epithelial Barrier Integrity and Inflammation-Related Gene Expression in the Colon
3.6. Transfer of FS–ASD in Mice Modulates Expression of Glial and Neuronal Molecules
3.7. FS Can Exert a Direct Effect on the ENS by Modulating Expression of Neuronal Molecules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Leventhal, B.L. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 2015, 77, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevison, C.; Blaxill, M.; Zahorodny, W. California Autism Prevalence Trends from 1931 to 2014 and Comparison to National ASD Data from IDEA and ADDM. J. Autism Dev. Disord. 2018, 48, 4103–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Albis, M.A.; Guevara, P.; Guevara, M.; Laidi, C.; Boisgontier, J.; Sarrazin, S.; Duclap, D.; Delorme, R.; Bolognani, F.; Czech, C.; et al. Local structural connectivity is associated with social cognition in autism spectrum disorder. Brain 2018, 141, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5(-)(/+)or EHMT2(-)(/+)human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. ELife 2019, 8. [Google Scholar] [CrossRef]
- Holiga, S.; Hipp, J.F.; Chatham, C.H.; Garces, P.; Spooren, W.; D’Ardhuy, X.L.; Bertolino, A.; Bouquet, C.; Buitelaar, J.K.; Bours, C.; et al. Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Zikopoulos, B.; Barbas, H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 2013, 7, 609. [Google Scholar] [CrossRef] [Green Version]
- Gillberg, C.; Billstedt, E. Autism and Asperger syndrome: Coexistence with other clinical disorders. Acta Psychiatr. Scand. 2000, 102, 321–330. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal symptoms in autism spectrum disorder: A meta-analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, N.; Chandler, S.; Turner, C.; Charman, T.; Pickles, A.; Loucas, T.; Simonoff, E.; Sullivan, P.; Baird, G. Gut permeability in autism spectrum disorders. Autism Res. Off. J. Int. Soc. Autism Res. 2014, 7, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Magistris, L.; Familiari, V.; Pascotto, A.; Sapone, A.; Frolli, A.; Iardino, P.; Carteni, M.; De Rosa, M.; Francavilla, R.; Riegler, G.; et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 418–424. [Google Scholar] [CrossRef] [Green Version]
- D’Eufemia, P.; Celli, M.; Finocchiaro, R.; Pacifico, L.; Viozzi, L.; Zaccagnini, M.; Cardi, E.; Giardini, O. Abnormal intestinal permeability in children with autism. Acta Paediatr. 1996, 85, 1076–1079. [Google Scholar] [CrossRef]
- Kushak, R.I.; Buie, T.M.; Murray, K.F.; Newburg, D.S.; Chen, C.; Nestoridi, E.; Winter, H.S. Evaluation of Intestinal Function in Children With Autism and Gastrointestinal Symptoms. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 687–691. [Google Scholar] [CrossRef]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 2016, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Gorrindo, P.; Williams, K.C.; Lee, E.B.; Walker, L.S.; McGrew, S.G.; Levitt, P. Gastrointestinal dysfunction in autism: Parental report, clinical evaluation, and associated factors. Autism Res. Off. J. Int. Soc. Autism Res. 2012, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 2018, 49, 121–131. [Google Scholar] [CrossRef]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Perez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [PubMed] [Green Version]
- Hosie, S.; Ellis, M.; Swaminathan, M.; Ramalhosa, F.; Seger, G.O.; Balasuriya, G.K.; Gillberg, C.; Rastam, M.; Churilov, L.; McKeown, S.J.; et al. Gastrointestinal dysfunction in patients and mice expressing the autism-associated R451C mutation in neuroligin-3. Autism Res. Off. J. Int. Soc. Autism Res. 2019, 12, 1043–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Dothel, G.; Barbaro, M.R.; Boudin, H.; Vasina, V.; Cremon, C.; Gargano, L.; Bellacosa, L.; De Giorgio, R.; Le Berre-Scoul, C.; Aubert, P.; et al. Nerve fiber outgrowth is increased in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology 2015, 148, 1002–1011. [Google Scholar] [CrossRef]
- Sasselli, V.; Boesmans, W.; Vanden Berghe, P.; Tissir, F.; Goffinet, A.M.; Pachnis, V. Planar cell polarity genes control the connectivity of enteric neurons. J. Clin. Investig. 2013, 123, 1763–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Silfhout, A.T.V.-v.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, K.G.; Li, Z.; Stevanovic, K.; Saurman, V.; Israelyan, N.; Anderson, G.M.; Snyder, I.; Veenstra-VanderWeele, J.; Blakely, R.D.; Gershon, M.D.; et al. Serotonin transporter variant drives preventable gastrointestinal abnormalities in development and function. J. Clin. Investig. 2016, 126, 2221–2235. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G.; Dunlop, S.A.; Mastaglia, F.L.; Akkari, P.A.; Koengten, F.; et al. Altered Gut Microbiome in Parkinson’s Disease and the Influence of Lipopolysaccharide in a Human α-Synuclein Over-Expressing Mouse Model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.G.; Kim, N.; Ju, I.G.; Eo, H.; Lim, S.M.; Jang, S.E.; Kim, D.H.; Oh, M.S. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 2018, 8, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef]
- Gondalia, S.V.; Palombo, E.A.; Knowles, S.R.; Cox, S.B.; Meyer, D.; Austin, D.W. Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res. Off. J. Int. Soc. Autism Res. 2012, 5, 419–427. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Elevated fecal short chain fatty acid and ammonia concentrations in children with autism spectrum disorder. Dig. Dis. Sci. 2012, 57, 2096–2102. [Google Scholar] [CrossRef]
- Iglesias-Vazquez, L.; Van Ginkel Riba, G.; Arija, V.; Canals, J. Composition of Gut Microbiota in Children with Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Li, J.; Wu, F.; Zheng, H.; Peng, Q.; Zhou, H. Altered composition and function of intestinal microbiota in autism spectrum disorders: A systematic review. Transl. Psychiatry 2019, 9, 43. [Google Scholar] [CrossRef]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2020, 319, G541–G548. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Yan, J.; Yang, T.; Zhu, J.; Li, T.; Wei, H.; Chen, J. Fecal Microbiome Transplantation from Children with Autism Spectrum Disorder Modulates Tryptophan and Serotonergic Synapse Metabolism and Induces Altered Behaviors in Germ-Free Mice. Msystems 2021, 6. [Google Scholar] [CrossRef]
- Foong, J.P.P.; Hung, L.Y.; Poon, S.; Savidge, T.C.; Bornstein, J.C. Early life interaction between the microbiota and the enteric nervous system. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G541–G548. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Borojevic, R.; Verdu, E.F.; Huizinga, J.D.; Ratcliffe, E.M. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol. Motil. 2014, 26, 98–107. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Grasset, E.; Manneras Holm, L.; Karsenty, G.; Macpherson, A.J.; Olofsson, L.E.; Backhed, F. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl. Acad. Sci. USA 2018, 115, 6458–6463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron 2015, 85, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Obata, Y.; Castano, A.; Boeing, S.; Bon-Frauches, A.C.; Fung, C.; Fallesen, T.; de Aguero, M.G.; Yilmaz, B.; Lopes, R.; Huseynova, A.; et al. Neuronal programming by microbiota regulates intestinal physiology. Nature 2020, 578, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Cossais, F.; Durand, T.; Chevalier, J.; Boudaud, M.; Kermarrec, L.; Aubert, P.; Neveu, I.; Naveilhan, P.; Neunlist, M. Postnatal development of the myenteric glial network and its modulation by butyrate. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G941–G951. [Google Scholar] [CrossRef] [Green Version]
- Suply, E.; de Vries, P.; Soret, R.; Cossais, F.; Neunlist, M. Butyrate enemas enhance both cholinergic and nitrergic phenotype of myenteric neurons and neuromuscular transmission in newborn rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1373–G1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquenlorge, S.; Duchalais, E.; Chevalier, J.; Cossais, F.; Rolli-Derkinderen, M.; Neunlist, M. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflamm. 2014, 11, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Rahnavard, A.; Chatterjee, S.; Sayoldin, B.; Crandall, K.A.; Tekola-Ayele, F.; Mallick, H. Omics community detection using multi-resolution clustering. Bioinformatics 2021. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.G.P.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120. [Google Scholar] [CrossRef]
- Trenteseaux, C.; Gaston, A.T.; Aguesse, A.; Poupeau, G.; de Coppet, P.; Andriantsitohaina, R.; Laschet, J.; Amarger, V.; Krempf, M.; Nobecourt-Dupuy, E.; et al. Perinatal Hypercholesterolemia Exacerbates Atherosclerosis Lesions in Offspring by Altering Metabolism of Trimethylamine-N-Oxide and Bile Acids. Arter. Thromb. Vasc. Biol. 2017, 37, 2053–2063. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, J.; Derkinderen, P.; Gomes, P.; Thinard, R.; Naveilhan, P.; Vanden Berghe, P.; Neunlist, M. Activity-dependent regulation of tyrosine hydroxylase expression in the enteric nervous system. J. Physiol. 2008, 586, 1963–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; Labaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Li, Q.; Zhang, J.; Wen, F.; Dang, W.; Duan, G.; Li, H.; Ruan, W.; Yang, P.; Guan, C.; et al. Characterization of Intestinal Microbiota and Probiotics Treatment in Children With Autism Spectrum Disorders in China. Front. Neurol. 2019, 10, 1084. [Google Scholar] [CrossRef] [Green Version]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabro, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal microbiota in children with autism in Slovakia. Physiol. Behav. 2015, 138, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Parracho, H.M.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rose, S.; Slattery, J.; MacFabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef] [PubMed]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23, 19620. [Google Scholar] [CrossRef]
- Thomas, R.H.; Meeking, M.M.; Mepham, J.R.; Tichenoff, L.; Possmayer, F.; Liu, S.; MacFabe, D.F. The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: Further development of a rodent model of autism spectrum disorders. J. Neuroinflammation 2012, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, J.; Chen, H.; Li, Y.; Amakye, W.K.; Liang, J.; Ma, B.; Chu, X.; Mao, L.; Zhang, Z. Fecal Short-Chain Fatty Acids Levels Were Not Associated With Autism Spectrum Disorders in Chinese Children: A Case-Control Study. Front. Neurosci. 2019, 13, 1216. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wu, B.; Zhang, Z.; Lu, H.; Fan, C.; Qi, Q.; Gao, Y.; Li, H.; Feng, C.; Zuo, J.; et al. Heme protects intestinal mucosal barrier in DSS-induced colitis through regulating macrophage polarization in both HO-1-dependent and HO-1-independent way. FASEB J. 2020, 34, 8028–8043. [Google Scholar] [CrossRef] [Green Version]
- Al-Sadi, R.; Ye, D.; Dokladny, K.; Ma, T.Y. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J. Immunol. 2008, 180, 5653–5661. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Z.; Liu, B.; Jin, Y.; Tian, Y.; Xin, Y.; Duan, Z. The Protective Effect of Heme Oxygenase-1 against Intestinal Barrier Dysfunction in Cholestatic Liver Injury Is Associated with NF-kappaB Inhibition. Mol. Med. 2017, 23, 215–224. [Google Scholar] [CrossRef]
- Raya-Sandino, A.; Castillo-Kauil, A.; Dominguez-Calderon, A.; Alarcon, L.; Flores-Benitez, D.; Cuellar-Perez, F.; Lopez-Bayghen, B.; Chavez-Munguia, B.; Vazquez-Prado, J.; Gonzalez-Mariscal, L. Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1714–1733. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Schneider, Y.; Brun, P.; Castagliuolo, I.; Pizzuti, D.; Martines, D.; Jond, L.; Bongiovanni, M.; Citi, S. Cingulin is dispensable for epithelial barrier function and tight junction structure, and plays a role in the control of claudin-2 expression and response to duodenal mucosa injury. J. Cell Sci. 2012, 125, 5005–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, T.; Torisawa, T.; Oiwa, K.; Tsukita, S. AMPK-dependent phosphorylation of cingulin reversibly regulates its binding to actin filaments and microtubules. Sci. Rep. 2018, 8, 15550. [Google Scholar] [CrossRef]
- Nava, P.; Capaldo, C.T.; Koch, S.; Kolegraff, K.; Rankin, C.R.; Farkas, A.E.; Feasel, M.E.; Li, L.; Addis, C.; Parkos, C.A.; et al. JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep. 2011, 12, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hao, M.M.; Van den Haute, C.; Baekelandt, V.; Boesmans, W.; Vanden Berghe, P. Regional complexity in enteric neuron wiring reflects diversity of motility patterns in the mouse large intestine. Elife 2019, 8. [Google Scholar] [CrossRef]
- Le Berre-Scoul, C.; Chevalier, J.; Oleynikova, E.; Cossais, F.; Talon, S.; Neunlist, M.; Boudin, H. A novel enteric neuron-glia coculture system reveals the role of glia in neuronal development. J. Physiol. 2017, 595, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Chen, Y.; Page, D.T. Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun. 2016, 7, 13421. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC | (n) | ASD | (n) | p Values | |
|---|---|---|---|---|---|
| SCFA (mM) | |||||
| Acetate | 6.57 ± 0.61 | (15) | 7.36 ± 0.47 | (38) | p = 0.356 |
| Propionate | 2.35 ± 0.28 | (15) | 2.57 ± 0.19 | (37) | p = 0.494 |
| Butyrate | 1.98 ± 0.19 | (15) | 2.61 ± 0.25 | (38) | p = 0.276 |
| Isobutyrate | 0.29 ± 0.04 | (15) | 0.31 ± 0.03 | (38) | p = 0.715 |
| Valerate | 0.26 ± 0.02 | (14) | 0.30 ± 0.02 | (38) | p = 0.688 |
| Bile acids (µg/mL) | |||||
| Primary | |||||
| CDCA | 0.045 ± 0.011 | (13) | 0.050 ± 0.007 | (37) | p = 0.747 |
| CA | 0.006 ± 0.002 | (13) | 0.009 ± 0.002 | (32) | p = 0.492 |
| Secondary | |||||
| DCA | 14.83 ± 4.24 | (13) | 26.87 ± 3.57 | (37) | p = 0.057 |
| LCA | 0.710 ± 0.162 | (12) | 0.537 ± 0.075 | (30) | p = 0.466 |
| UDCA | 0.030 ± 0.007 | (12) | 0.043 ± 0.009 | (25) | p = 0.471 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzales, J.; Marchix, J.; Aymeric, L.; Le Berre-Scoul, C.; Zoppi, J.; Bordron, P.; Burel, M.; Davidovic, L.; Richard, J.-R.; Gaman, A.; et al. Fecal Supernatant from Adult with Autism Spectrum Disorder Alters Digestive Functions, Intestinal Epithelial Barrier, and Enteric Nervous System. Microorganisms 2021, 9, 1723. https://doi.org/10.3390/microorganisms9081723
Gonzales J, Marchix J, Aymeric L, Le Berre-Scoul C, Zoppi J, Bordron P, Burel M, Davidovic L, Richard J-R, Gaman A, et al. Fecal Supernatant from Adult with Autism Spectrum Disorder Alters Digestive Functions, Intestinal Epithelial Barrier, and Enteric Nervous System. Microorganisms. 2021; 9(8):1723. https://doi.org/10.3390/microorganisms9081723
Chicago/Turabian StyleGonzales, Jacques, Justine Marchix, Laetitia Aymeric, Catherine Le Berre-Scoul, Johanna Zoppi, Philippe Bordron, Marie Burel, Laetitia Davidovic, Jean-Romain Richard, Alexandru Gaman, and et al. 2021. "Fecal Supernatant from Adult with Autism Spectrum Disorder Alters Digestive Functions, Intestinal Epithelial Barrier, and Enteric Nervous System" Microorganisms 9, no. 8: 1723. https://doi.org/10.3390/microorganisms9081723