
Characterizing the Cattle Gut Microbiome in Farms with a High and Low Prevalence of Shiga Toxin Producing Escherichia coli

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Pathogen Identification
2.3. Amplicon Library Processing
2.4. Microbiome Analyses
3. Results
3.1. Farm Characteristics
3.2. Sequencing Results
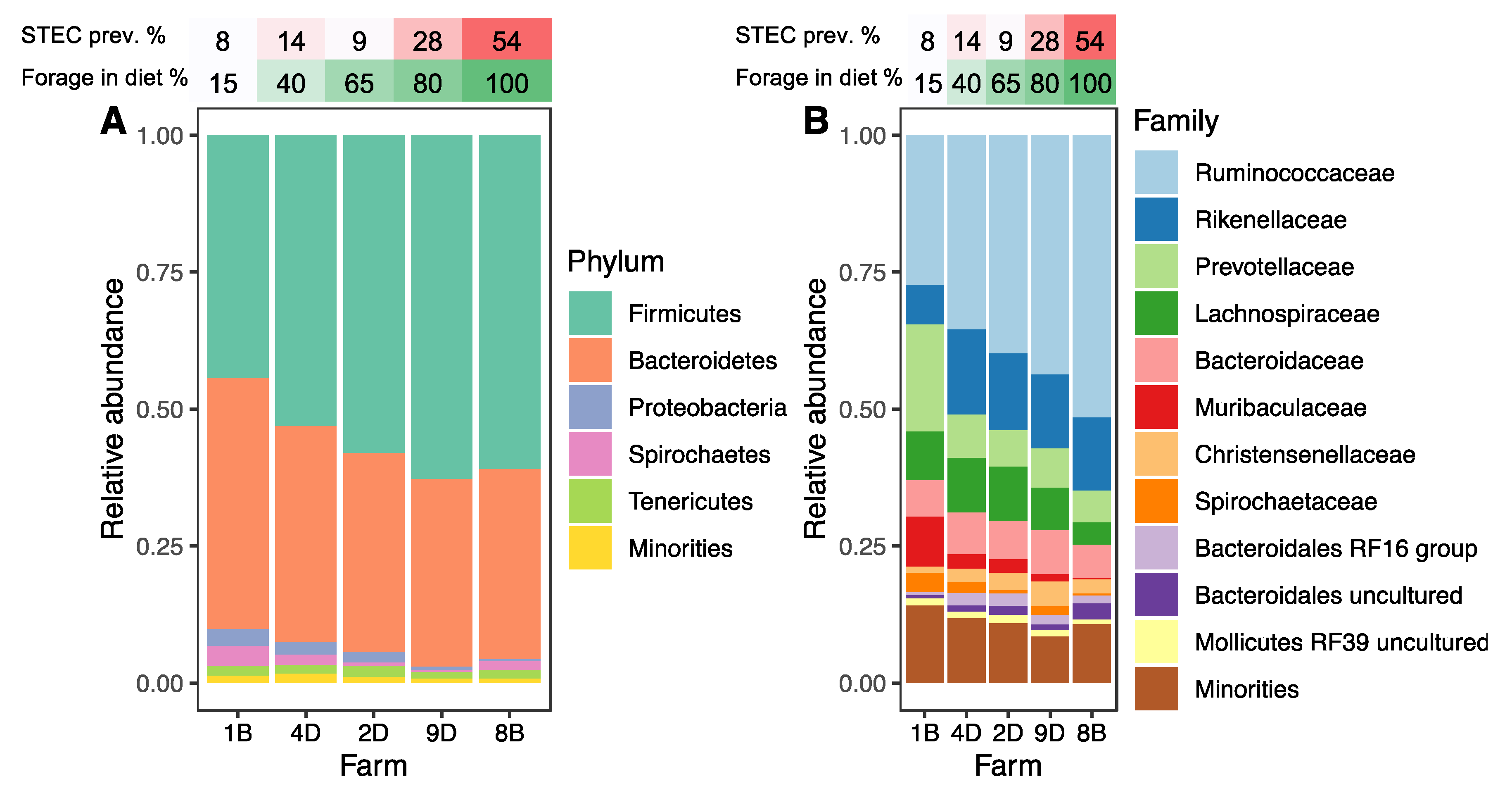
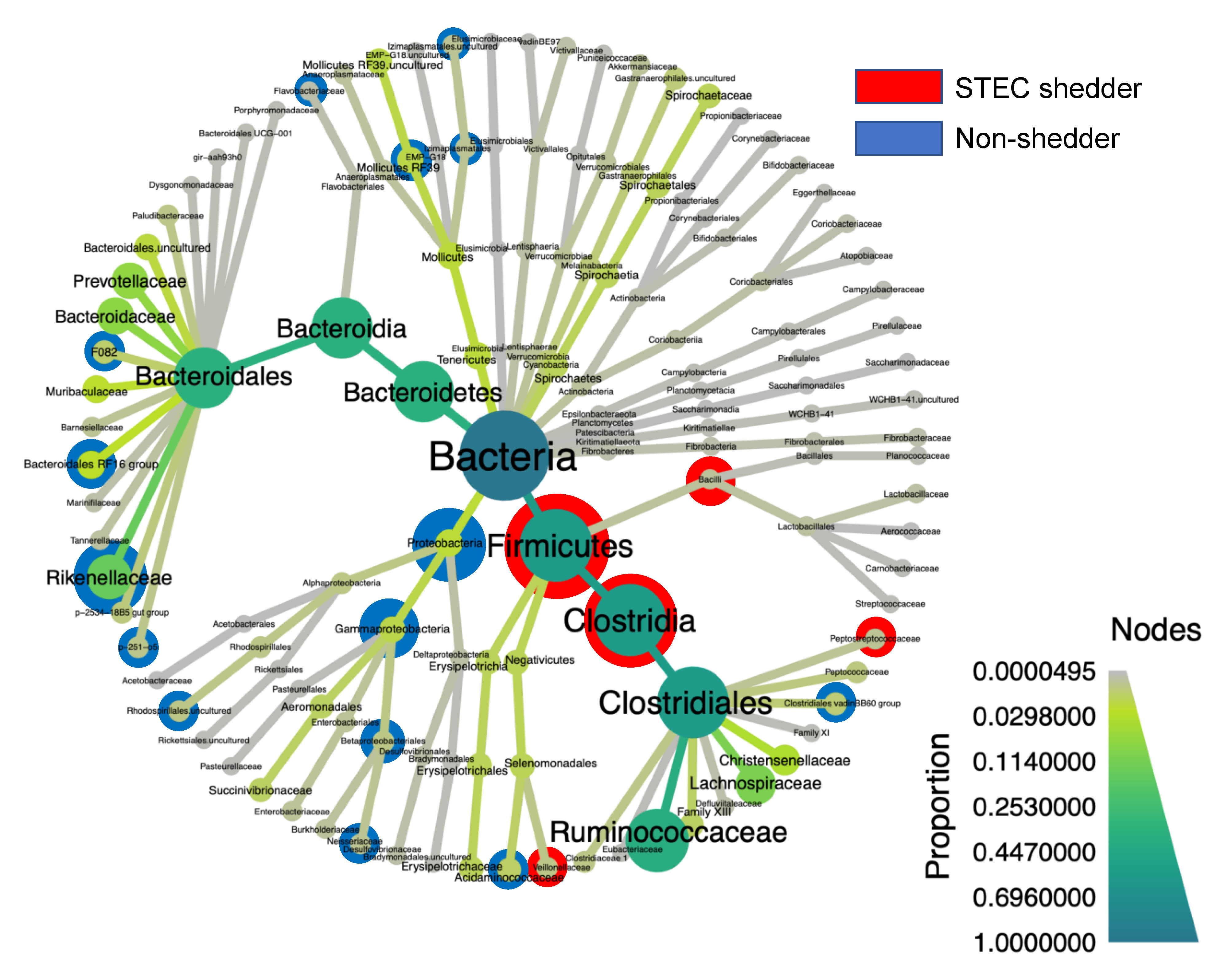
3.3. Hindgut Microbiota Composition
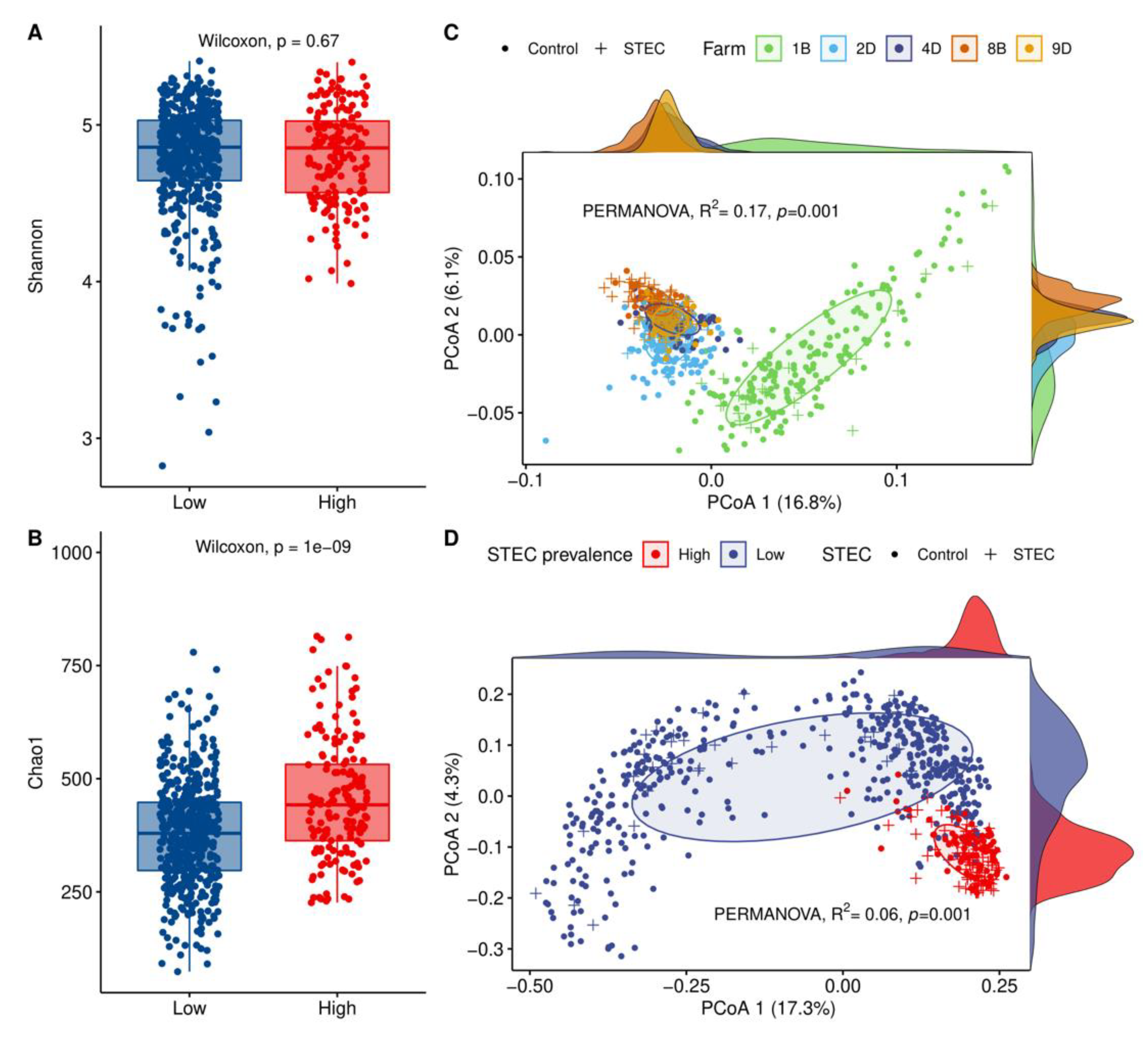
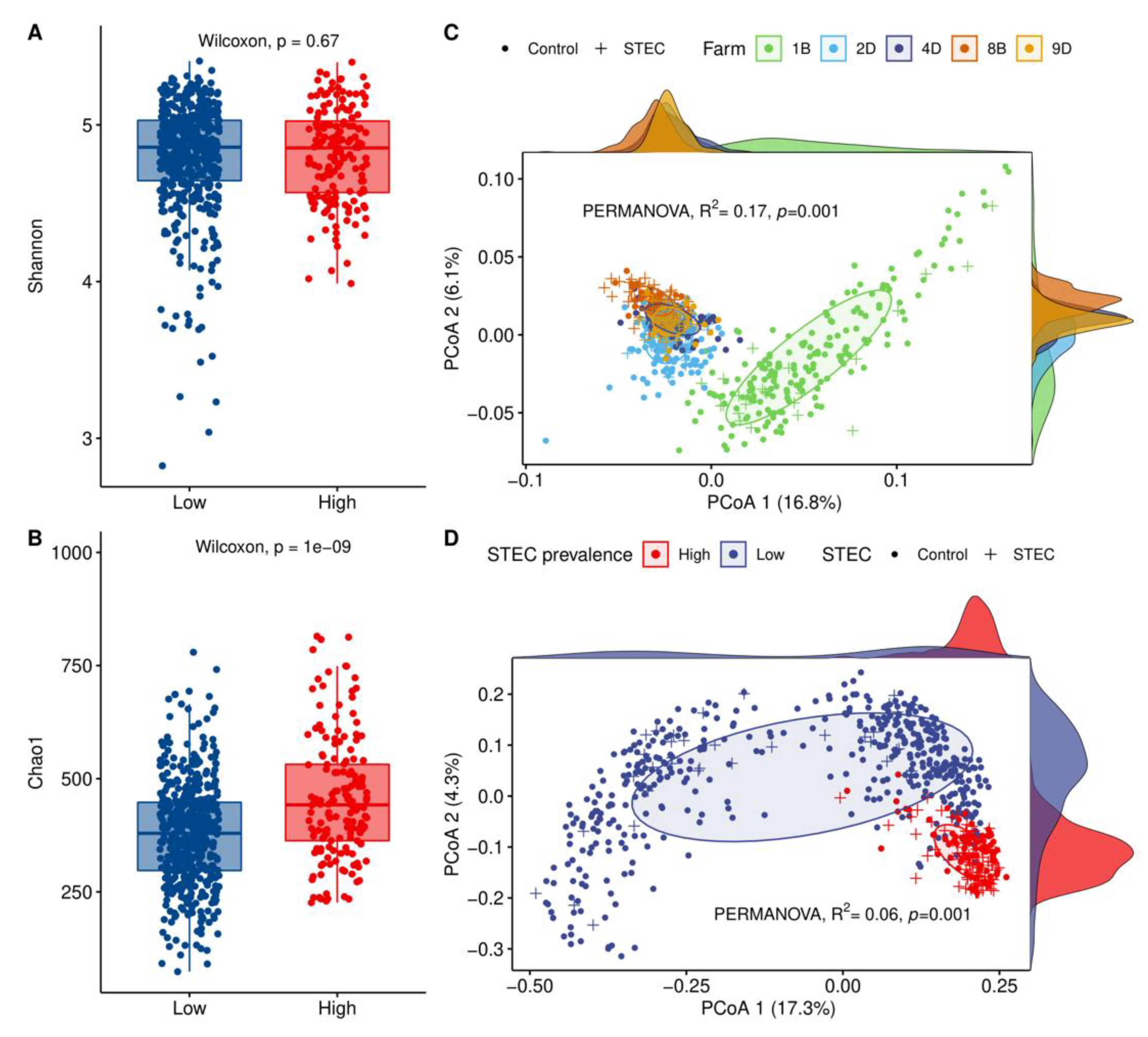
3.4. HSP Farms Characterized by Forage-Dominant Diets Exhibited Higher Alpha Diversity and a Distinct Microbiota Structure
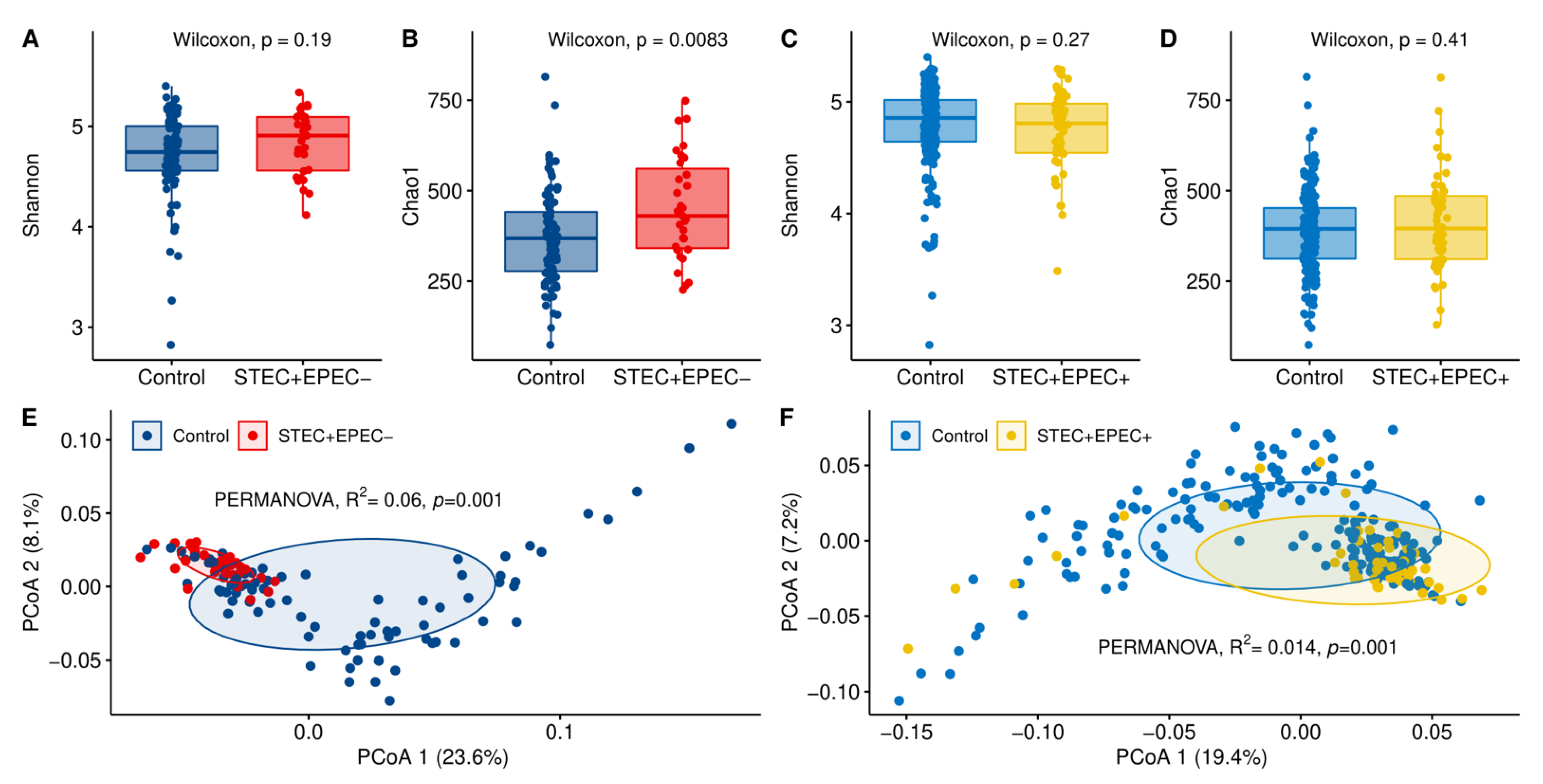
3.5. Hindgut Microbiota Diversity Comparisons between STEC-Positive Samples and Controls
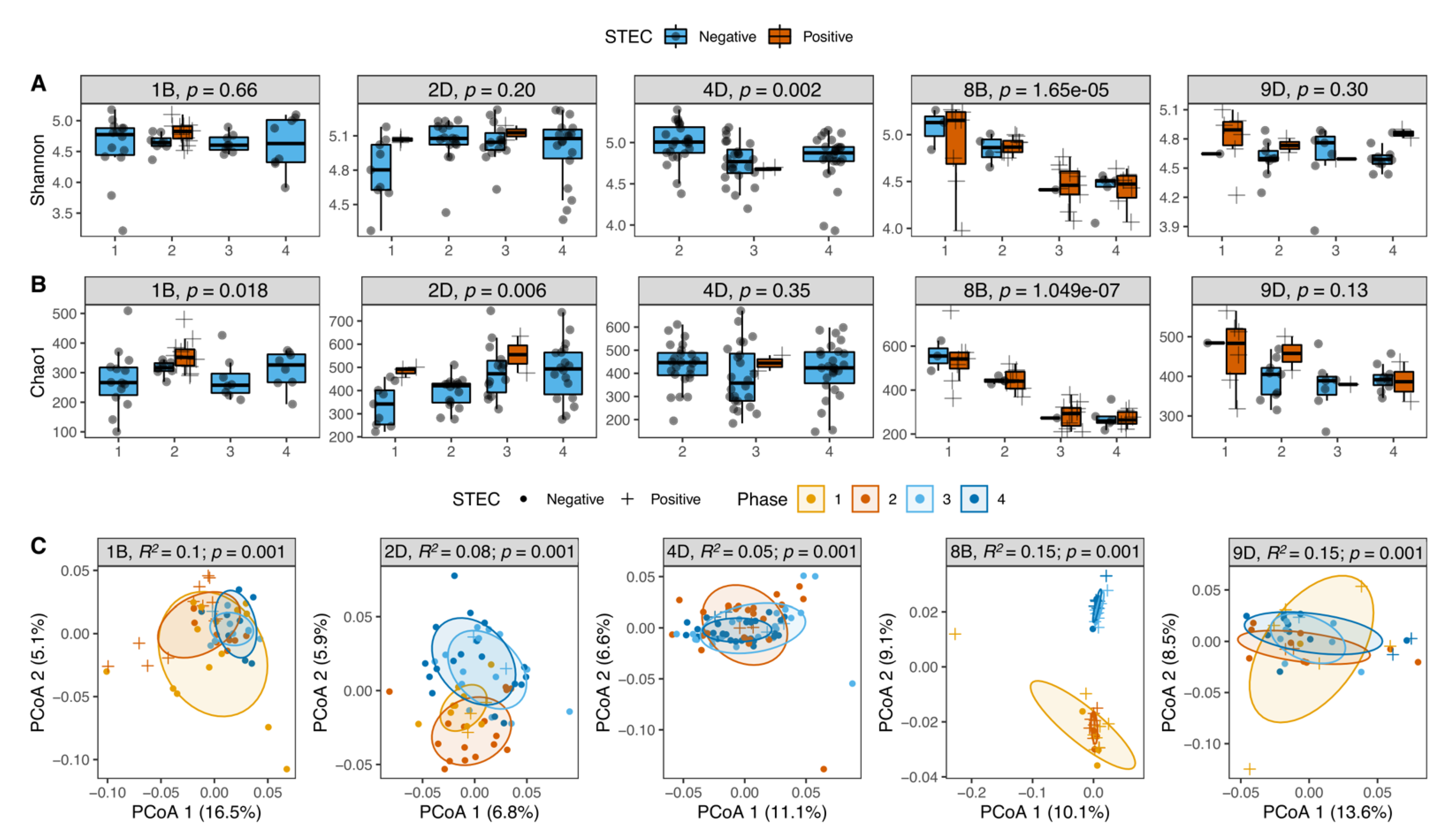
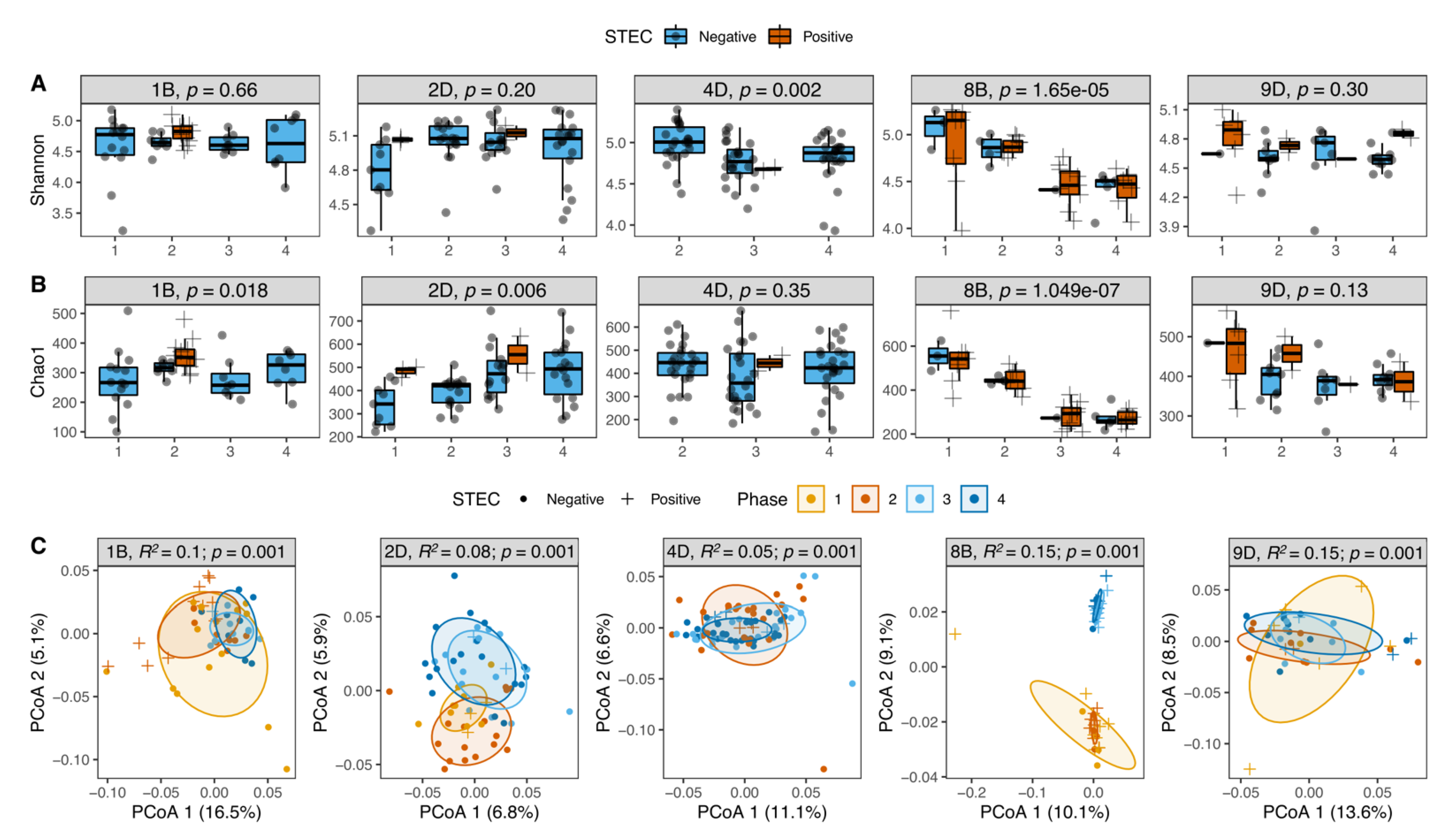
3.6. STEC Carriers from Farms with LSP but Not HSP Showed Changes in Microbiota Diversity over Time
3.7. Differentially Abundant Taxa among the STEC Shedders from Dairy Farms
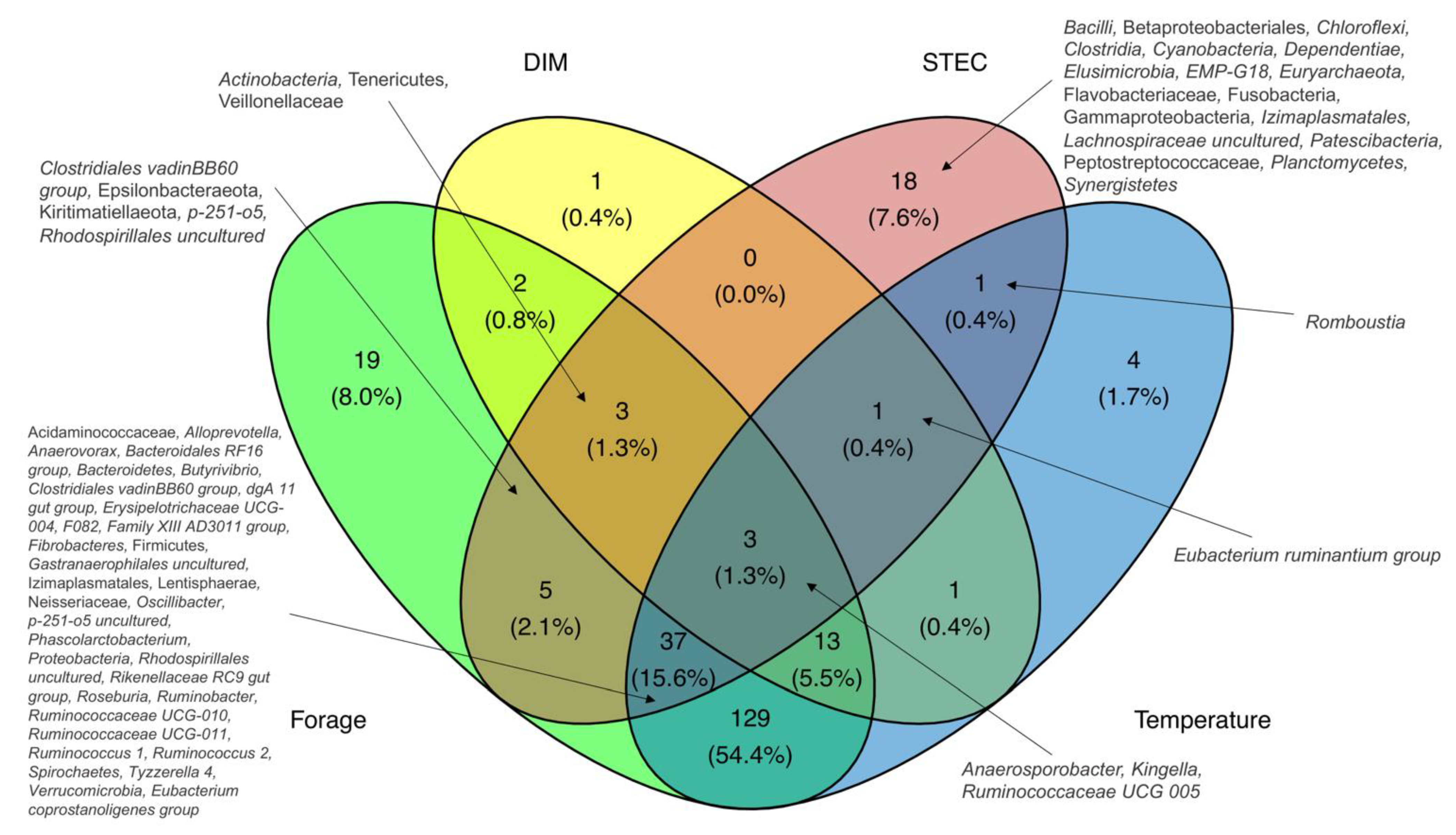
3.8. Taxa Correlated with Factors Associated with STEC Carriage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Dairy Farm | ||
|---|---|---|---|
| 2D | 4D | 9D | |
| No. of milkings/day | 2 times | 3 times | 2–3 times |
| DIM (SD) | 206.7 (134) | 259 (118.5) | 195.25 (136.43) |
| No. of lactations | |||
| 0 (No. of cows) | 4 | 0 | 0 |
| 1 (No. of cows) | 80 | 36 | 34 |
| ≥2 (No. of cows) | 141 | 86 | 43 |
| Dry (No. of cows) | 5 | 9 | 0 |
| Treatment | |||
| Clinical mastitis | Ceftiofur, pirlimycin hydrochloride, penicillin, ampicillin, oxytetracycline, sulfadimethoxine | Penicillin G procaine, ceftiofur, pirlimycin hydrochloride, amoxicillin | Ceftiofur |
| Metritis | Oxytetracycline, penicillin | Ceftiofur | Ceftiofur, isoflupredone acetate |
| Dry | Penicillin-novobiocin, Penicillin-dihydrostreptomycin | Penicillin-dihydrostreptomycin, Orbeseal | None |
References
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global Incidence of Human Shiga Toxin–Producing Escherichia coli Infections and Deaths: A Systematic Review and Knowledge Synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapountzis, P.; Segura, A.; Desvaux, M.; Forano, E. An Overview of the Elusive Passenger in the Gastrointestinal Tract of Cattle: The Shiga Toxin Producing Escherichia coli. Microorganisms 2020, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in Bacteriophage Lambda Development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Pierard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter Evaluation of a Sequence-Based Protocol for Subtyping Shiga Toxins and Standardizing Stx Nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, A.D.; Holmes, R.K. Shiga and Shiga-like Toxins. Microbiol. Rev. 1987, 51, 206–220. [Google Scholar] [CrossRef]
- O’Brien, A.; Newland, J.; Miller, S.; Holmes, R.; Smith, H.; Formal, S. Shiga-like Toxin-Converting Phages from Escherichia coli Strains That Cause Hemorrhagic Colitis or Infantile Diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef]
- Pruimboom-Brees, I.M.; Morgan, T.W.; Ackermann, M.R.; Nystrom, E.D.; Samuel, J.E.; Cornick, N.A.; Moon, H.W. Cattle Lack Vascular Receptors for Escherichia coli O157:H7 Shiga Toxins. Proc. Natl. Acad. Sci. USA 2000, 97, 10325–10329. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, T.K.; Kaper, J.B. A Cloned Pathogenicity Island from Enteropathogenic Escherichia coli Confers the Attaching and Effacing Phenotype on E. coli K-12. Mol. Microbiol. 1997, 23, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Naylor, S.W.; Low, J.C.; Besser, T.E.; Mahajan, A.; Gunn, G.J.; Pearce, M.C.; McKendrick, I.J.; Smith, D.G.E.; Gally, D.L. Lymphoid Follicle-Dense Mucosa at the Terminal Rectum Is the Principal Site of Colonization of Enterohemorrhagic Escherichia coli O157:H7 in the Bovine Host. Infect. Immun. 2003, 71, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.E.; Elliott, E.J. Interventions for Preventing Diarrhea-Associated Hemolytic Uremic Syndrome: Systematic Review. BMC Public Health 2013, 13, 799. [Google Scholar] [CrossRef] [Green Version]
- Venegas-Vargas, C.; Henderson, S.; Khare, A.; Mosci, R.E.; Lehnert, J.D.; Singh, P.; Ouellette, L.M.; Norby, B.; Funk, J.A.; Rust, S.; et al. Factors Associated with Shiga Toxin-Producing Escherichia coli Shedding by Dairy and Beef Cattle. Appl. Environ. Microbiol. 2016, 82, 5049–5056. [Google Scholar] [CrossRef] [Green Version]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of Pathogens and Pathobionts by the Gut Microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Gigliucci, F.; von Meijenfeldt, F.A.B.; Knijn, A.; Michelacci, V.; Scavia, G.; Minelli, F.; Dutilh, B.E.; Ahmad, H.M.; Raangs, G.C.; Friedrich, A.W.; et al. Metagenomic Characterization of the Human Intestinal Microbiota in Fecal Samples from STEC-Infected Patients. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Teal, T.K.; Marsh, T.L.; Tiedje, J.M.; Mosci, R.; Jernigan, K.; Zell, A.; Newton, D.W.; Salimnia, H.; Lephart, P.; et al. Intestinal Microbial Communities Associated with Acute Enteric Infections and Disease Recovery. Microbiome 2015, 3, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-S.; Jeong, Y.-J.; Lee, M.-S. Escherichia coli Shiga Toxins and Gut Microbiota Interactions. Toxins 2021, 13, 416. [Google Scholar] [CrossRef]
- Xu, Y.; Dugat-Bony, E.; Zaheer, R.; Selinger, L.; Barbieri, R.; Munns, K.; McAllister, T.A.; Selinger, L.B. Escherichia coli O157:H7 Super-Shedder and Non-Shedder Feedlot Steers Harbour Distinct Fecal Bacterial Communities. PLoS ONE 2014, 9, e98115. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Tyler, P.J.; Starnes, J.; Bratcher, C.L.; Rankins, D.; McCaskey, T.A.; Wang, L. Correlation Analysis of Shiga Toxin-Producing Escherichia coli Shedding and Faecal Bacterial Composition in Beef Cattle. J. Appl. Microbiol. 2013, 115, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.-J.; Kim, W.; An, J.-U.; Kim, J.; Cho, S. The Fecal Microbial Communities of Dairy Cattle Shedding Shiga Toxin–Producing Escherichia coli or Campylobacter jejuni. Foodborne Pathog. Dis. 2016, 13, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Salaheen, S.; Kim, S.W.; Karns, J.S.; Hovingh, E.; Haley, B.J.; Van Kessel, J.A.S. Metagenomic Analysis of the Fecal Microbiomes from Escherichia coli O157:H7-Shedding and Non-Shedding Cows on a Single Dairy Farm. Food Control 2019, 102, 76–80. [Google Scholar] [CrossRef]
- Mir, R.A.; Weppelmann, T.A.; Elzo, M.; Ahn, S.; Driver, J.D.; Jeong, K.C. Colonization of Beef Cattle by Shiga Toxin-Producing Escherichia coli during the First Year of Life: A Cohort Study. PLoS ONE 2016, 11, e0148518. [Google Scholar] [CrossRef] [Green Version]
- Wang, O.; McAllister, T.A.; Plastow, G.; Stanford, K.; Selinger, B.; Guan, L.L. Interactions of the Hindgut Mucosa-Associated Microbiome with Its Host Regulate Shedding of Escherichia coli O157:H7 by Cattle. Appl. Environ. Microbiol. 2017, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.A.; Schaut, R.G.; Looft, T.; Allen, H.K.; Sharma, V.K.; Kudva, I.T. Recto-Anal Junction (RAJ) and Fecal Microbiomes of Cattle Experimentally Challenged With Escherichia coli O157:H7. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Stenkamp-Strahm, C.; McConnel, C.; Magzamen, S.; Abdo, Z.; Reynolds, S. Associations between Escherichia coli O157 Shedding and the Faecal Microbiota of Dairy Cows. J. Appl. Microbiol. 2018, 124, 881–898. [Google Scholar] [CrossRef]
- Kim, M.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. The Impact of the Bovine Faecal Microbiome on Escherichia coli O157:H7 Prevalence and Enumeration in Naturally Infected Cattle. J. Appl. Microbiol. 2017, 123, 1027–1042. [Google Scholar] [CrossRef]
- Venegas-Vargas, C.; Manning, S.D.; Coussens, P.M.; Roussey, J.A.; Bartlett, P.; Grooms, D. Bovine Leukemia Virus and Mycobacterium avium subsp. paratuberculosis Are Not Associated with Shiga Toxin–Producing Escherichia coli Shedding in Cattle. J. Food Prot. 2017, 80, 86–89. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2013, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeder, J.; Knight, R. Rapidly Denoising Pyrosequencing Amplicon Reads by Exploiting Rank-Abundance Distributions. Nat. Methods 2010, 7, 668–669. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A Flexible Tool for Aligning Sequences to a Template Alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Mirarab, S.; Nguyen, N.; Warnow, T. SEPP: SATé-Enabled Phylogenetic Placement. In Proceedings of the Biocomputing 2012; World Scientific: Singapore, 2011; pp. 247–258. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; et al. Package ‘Vegan’ Version 2.5-6. Community Ecol. Package Version 2019, 2, 1–295. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, J.; Murakami, H.; Sato, R.; Mizukami, K.; Suzuki, T.; Shima, A.; Ishihara, G.; Sogawa, K.; Sakaguchi, M. Examination of the Fecal Microbiota in Dairy Cows Infected with Bovine Leukemia Virus. Vet. Microbiol. 2020, 240, 108547. [Google Scholar] [CrossRef]
- Fecteau, M.-E.; Pitta, D.W.; Vecchiarelli, B.; Indugu, N.; Kumar, S.; Gallagher, S.C.; Fyock, T.L.; Sweeney, R.W. Dysbiosis of the Fecal Microbiota in Cattle Infected with Mycobacterium avium subsp. paratuberculosis. PLoS ONE 2016, 11, e0160353. [Google Scholar] [CrossRef]
- Hagey, J.V.; Bhatnagar, S.; Heguy, J.M.; Karle, B.M.; Price, P.L.; Meyer, D.; Maga, E.A. Fecal Microbial Communities in a Large Representative Cohort of California Dairy Cows. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.L.; Bono, J.L.; DeSantis, T.Z.; Harhay, D.M.; Andersen, G.L.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-Animal Variation in Fecal Microbial Diversity among Beef Cattle. Appl. Environ. Microbiol. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shi, H.; Wang, Y.; Cao, Z.; Yang, H.; Li, S. Effect of Limit-Fed Diets With Different Forage to Concentrate Ratios on Fecal Bacterial and Archaeal Community Composition in Holstein Heifers. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Kim, M.; Kim, J.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. Investigation of Bacterial Diversity in the Feces of Cattle Fed Different Diets. J. Anim. Sci. 2014, 92, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, F.; Cai, W.; Jia, C.; Bai, Y.; He, Y.; Zhu, W.; Li, R.W.; Song, J. Diet-Induced Changes in Bacterial Communities in the Jejunum and Their Associations with Bile Acids in Angus Beef Cattle. Anim. Microbiome 2020, 2, 33. [Google Scholar] [CrossRef]
- Jacob, M.E.; Callaway, T.R.; Nagaraja, T.G. Dietary Interactions and Interventions Affecting Escherichia coli O157 Colonization and Shedding in Cattle. Foodborne Pathog. Dis. 2009, 6, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, T.R.; Carr, M.A.; Edrington, T.S.; Anderson, R.C.; Nisbet, D.J. Diet, Escherichia coli O157:H7, and Cattle: A Review after 10 Years. Curr. Issues Mol. Biol. 2009, 11, 67–79. [Google Scholar] [PubMed]
- Kunz, I.G.Z.; Reed, K.J.; Metcalf, J.L.; Hassel, D.M.; Coleman, R.J.; Hess, T.M.; Coleman, S.J. Equine Fecal Microbiota Changes Associated with Anthelmintic Administration. J. Equine Vet. Sci. 2019, 77, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, M.A.; Lidbury, J.A.; Pilla, R.; Steiner, J.M.; Lappin, M.R.; Suchodolski, J.S. Evaluation of the Effects of Anthelmintic Administration on the Fecal Microbiome of Healthy Dogs with and without Subclinical Giardia spp. and Cryptosporidium canis Infections. PLoS ONE 2020, 15, e0228145. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.V.; Quiñones, M.; Vujkovic-Cvijin, I.; Oliveira, R.G.; Kepha, S.; Odiere, M.R.; Anderson, R.M.; Belkaid, Y.; Nutman, T.B. The Impact of Anthelmintic Treatment on Human Gut Microbiota Based on Cross-Sectional and Pre- and Postdeworming Comparisons in Western Kenya. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Rosa, B.A.; Supali, T.; Gankpala, L.; Djuardi, Y.; Sartono, E.; Zhou, Y.; Fischer, K.; Martin, J.; Tyagi, R.; Bolay, F.K.; et al. Differential Human Gut Microbiome Assemblages during Soil-Transmitted Helminth Infections in Indonesia and Liberia. Microbiome 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, R.; Dugat-Bony, E.; Holman, D.; Cousteix, E.; Xu, Y.; Munns, K.; Selinger, L.J.; Barbieri, R.; Alexander, T.; McAllister, T.A.; et al. Changes in Bacterial Community Composition of Escherichia coli O157:H7 Super-Shedder Cattle Occur in the Lower Intestine. PLoS ONE 2017, 12, e0170050. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.A.; Schaut, R.G.; Allen, H.K.; Looft, T.; Loving, C.L.; Kudva, I.T.; Sharma, V.K. Cattle Intestinal Microbiota Shifts Following Escherichia coli O157:H7 Vaccination and Colonization. PLoS ONE 2019, 14, e0226099. [Google Scholar] [CrossRef]
- Blankenship, H.M.; Carbonell, S.; Mosci, R.; McWilliams, K.; Pietrzen, K.; Benko, S.; Gatesy, T.; Grooms, D.; Manning, S.D. Genetic and Phenotypic Factors Associated with Persistent Shiga Toxin-Producing Escherichia coli Shedding in Beef Cattle. Appl. Environ. Microbiol. 2020. [Google Scholar] [CrossRef]
- Chopyk, J.; Moore, R.M.; DiSpirito, Z.; Stromberg, Z.R.; Lewis, G.L.; Renter, D.G.; Cernicchiaro, N.; Moxley, R.A.; Wommack, K.E. Presence of Pathogenic Escherichia coli Is Correlated with Bacterial Community Diversity and Composition on Pre-Harvest Cattle Hides. Microbiome 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerritsen, J.; Umanets, A.; Staneva, I.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; de Vos, W.M.; Smidt, H. Romboutsia hominis sp. nov., the First Human Gut-Derived Representative of the Genus Romboutsia, Isolated from Ileostoma Effluent. Int. J. Syst. Evol. Microbiol. 2018, 68, 3479–3486. [Google Scholar] [CrossRef]
- Downes, J.; Dewhirst, F.E.; Tanner, A.C.R.; Wade, W.G.Y. 2013 Description of Alloprevotella rava gen. nov., sp. nov., Isolated from the Human Oral Cavity, and Reclassification of Prevotella tannerae Moore et al. 1994 as Alloprevotella tannerae gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2013, 63, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Krumholz, L.R.; Bryant, M.P.; Brulla, W.J.; Vicini, J.L.; Clark, J.H.; Stahl, D.A.Y. 1993 Proposal of Quinella ovalis gen. nov., sp. nov., Based on Phylogenetic Analysis. Int. J. Syst. Evol. Microbiol. 1993, 43, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Alou, M.T.; Ndongo, S.; Frégère, L.; Labas, N.; Andrieu, C.; Richez, M.; Couderc, C.; Baudoin, J.-P.; Abrahão, J.; Brah, S.; et al. Taxonogenomic Description of Four New Clostridium Species Isolated from Human Gut: “Clostridium amazonitimonense”, “Clostridium merdae”, “Clostridium massilidielmoense” and “Clostridium nigeriense”. New Microbes New Infect. 2018, 21, 128–139. [Google Scholar] [CrossRef]
- Brenner, D.J.; Krieg, N.; Staley, J.; Garrity, G. Bergey’s Manual of Systematic Bacteriology, Vol 2: The Proteobacteria; Springer: New York, NY, USA, 2005. [Google Scholar]
- Yagupsky, P. Kingella kingae: Carriage, Transmission, and Disease. Clin. Microbiol. Rev. 2015, 28, 54–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-Utilizing Capacity Varies in Prevotella- versus Bacteroides-Dominated Gut Microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin Study Indicates Loss of Interaction between Microbiota and Mucosa of Patients with Ulcerative Colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol Trisphosphate and Diacylglycerol as Intracellular Second Messengers. In Mechanisms of Receptor Regulation; Poste, G., Crooke, S.T., Eds.; Springer US: Boston, MA, USA, 1985; pp. 111–130. ISBN 978-1-4613-2131-6. [Google Scholar]
- Saleem, F.; Ametaj, B.N.; Bouatra, S.; Mandal, R.; Zebeli, Q.; Dunn, S.M.; Wishart, D.S. A Metabolomics Approach to Uncover the Effects of Grain Diets on Rumen Health in Dairy Cows. J. Dairy Sci. 2012, 95, 6606–6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertin, Y.; Girardeau, J.P.; Chaucheyras-Durand, F.; Lyan, B.; Pujos-Guillot, E.; Harel, J.; Martin, C. Enterohaemorrhagic Escherichia coli Gains a Competitive Advantage by Using Ethanolamine as a Nitrogen Source in the Bovine Intestinal Content. Environ. Microbiol. 2011, 13, 365–377. [Google Scholar] [CrossRef]
- Thiennimitr, P.; Winter, S.E.; Winter, M.G.; Xavier, M.N.; Tolstikov, V.; Huseby, D.L.; Sterzenbach, T.; Tsolis, R.M.; Roth, J.R.; Bäumler, A.J. Intestinal Inflammation Allows Salmonella to Use Ethanolamine to Compete with the Microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 17480–17485. [Google Scholar] [CrossRef] [Green Version]
- Snelling, T.J.; Auffret, M.D.; Duthie, C.-A.; Stewart, R.D.; Watson, M.; Dewhurst, R.J.; Roehe, R.; Walker, A.W. Temporal Stability of the Rumen Microbiota in Beef Cattle, and Response to Diet and Supplements. Anim. Microbiome 2019, 1, 16. [Google Scholar] [CrossRef]
- Murphy, B.P.; McCabe, E.; Murphy, M.; Buckley, J.F.; Crowley, D.; Fanning, S.; Duffy, G. Longitudinal Study of Two Irish Dairy Herds: Low Numbers of Shiga Toxin-Producing Escherichia coli O157 and O26 Super-Shedders Identified. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, R.N.; Hancock, D.D.; Rice, D.H.; Berg, J.; Stilborn, R.; Hovde, C.J.; Besser, T.E. Rectoanal Junction Colonization of Feedlot Cattle by Escherichia coli O157:H7 and Its Association with Supershedders and Excretion Dynamics. Appl. Environ. Microbiol. 2007, 73, 1563–1568. [Google Scholar] [CrossRef] [Green Version]
- Cernicchiaro, N.; Pearl, D.L.; McEwen, S.A.; Zerby, H.N.; Fluharty, F.L.; Loerch, S.C.; Kauffman, M.D.; Bard, J.L.; LeJeune, J.T. A Randomized Controlled Trial to Assess the Impact of Dietary Energy Sources, Feed Supplements, and the Presence of Super-Shedders on the Detection of Escherichia coli O157:H7 in Feedlot Cattle Using Different Diagnostic Procedures. Foodborne Pathog. Dis. 2010, 7, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Menrath, A.; Wieler, L.H.; Heidemanns, K.; Semmler, T.; Fruth, A.; Kemper, N. Shiga Toxin Producing Escherichia coli: Identification of Non-O157:H7-Super-Shedding Cows and Related Risk Factors. Gut Pathog. 2010, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]




| Feature | Farm | ||||
|---|---|---|---|---|---|
| 2D | 4D | 9D | 1B | 8B | |
| Breed | Holstein | Holstein | Holstein | Crossbreed | Angus |
| Herd | Dairy | Dairy | Dairy | Beef | Beef |
| Herd size | 320 | 3000 | 243 | 136 | 54 |
| STEC prevalence (%) | 8.7 | 13.8 | 28.0 | 8.2 | 53.7 |
| STEC prevalence classification | LSP | LSP | HSP | LSP | HSP |
| No. of samples | 213 | 81 | 77 | 206 | 83 |
| Fecal grab | 48 | 40 | 77 | 60 | 83 |
| Recto-anal junction | 165 | 41 | 0 | 146 | 0 |
| Mean age days (SD a) | 1382 (476) | NR b | 1362 (522) | 372 (19) | 442 (17) |
| Housing | Free stall; tie stall | Free stall | Access to pasture/dry lot; free stall | Feedlot | Loose house |
| Diet % (SD) | |||||
| Forage | 65.01 (18.76) | 40.62 (9.47) | 80 (0) | 15 (0) | 100 (0) |
| Concentrate | 34.99 (18.76) | 59.38 (9.47) | 20 (0) | 85 (0) | 0 |
| Corn silage | 29.06 (8.82) | 41.7 (3.74) | 0 | 15 (0) | 0 |
| Cotton seed | 1.60 (2.60) | 0 | 0 | 0 | 0 |
| Rumensin | No | Yes | No | Yes | No |
| Roughage, protein | No | No | Yes | No | No |
| Season c | Summer | Summer | Summer | Spring | Summer |
| Temperature (°C) | 25 | 25 | 36 | 4.4 | 36 |
| Humidity (g/m3) | 66 | 68 | 31 | 75 | 42 |
| Temp. max. 5 days d | 23.44 | 29.89 | 37.11 | 20.33 | 29.33 |
| Temp. avg. 5 days d | 19.22 | 16.89 | 30.11 | 13.89 | 22.78 |
| Treatment | |||||
| Anthelmintic | Yes | Yes | No | Yes | No |
| Respiratory | Ceftiofur, florfenicol | Ceftiofur | None | Ceftiofur, tulathromycin | Florfenicol |
| Foot infection | Copper sulfate, penicillin | Copper sulfate | Copper sulfate, oxytetracycline, ceftiofur | Oxytetracycline | Ceftiofur |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasco, K.; Nohomovich, B.; Singh, P.; Venegas-Vargas, C.; Mosci, R.E.; Rust, S.; Bartlett, P.; Norby, B.; Grooms, D.; Zhang, L.; et al. Characterizing the Cattle Gut Microbiome in Farms with a High and Low Prevalence of Shiga Toxin Producing Escherichia coli. Microorganisms 2021, 9, 1737. https://doi.org/10.3390/microorganisms9081737
Vasco K, Nohomovich B, Singh P, Venegas-Vargas C, Mosci RE, Rust S, Bartlett P, Norby B, Grooms D, Zhang L, et al. Characterizing the Cattle Gut Microbiome in Farms with a High and Low Prevalence of Shiga Toxin Producing Escherichia coli. Microorganisms. 2021; 9(8):1737. https://doi.org/10.3390/microorganisms9081737
Chicago/Turabian StyleVasco, Karla, Brian Nohomovich, Pallavi Singh, Cristina Venegas-Vargas, Rebekah E. Mosci, Steven Rust, Paul Bartlett, Bo Norby, Daniel Grooms, Lixin Zhang, and et al. 2021. "Characterizing the Cattle Gut Microbiome in Farms with a High and Low Prevalence of Shiga Toxin Producing Escherichia coli" Microorganisms 9, no. 8: 1737. https://doi.org/10.3390/microorganisms9081737