
Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations




Abstract
:1. Introduction
2. Materials and Methods
2.1. Nucleic Acid Extracts of Ticks and Metadata Management
2.2. PCR Amplification for Tick Samples and Bacterial 16S rRNA Gene Sequencing
2.3. Statistics
2.4. Analysis of Microbial Networks
3. Results
3.1. Presence of Human Pathogenic Bb Species Affected the Tick Microbiome Composition
3.2. Sequence Read Abundances of Wolbachia and Pseudomonas Differed between Locations
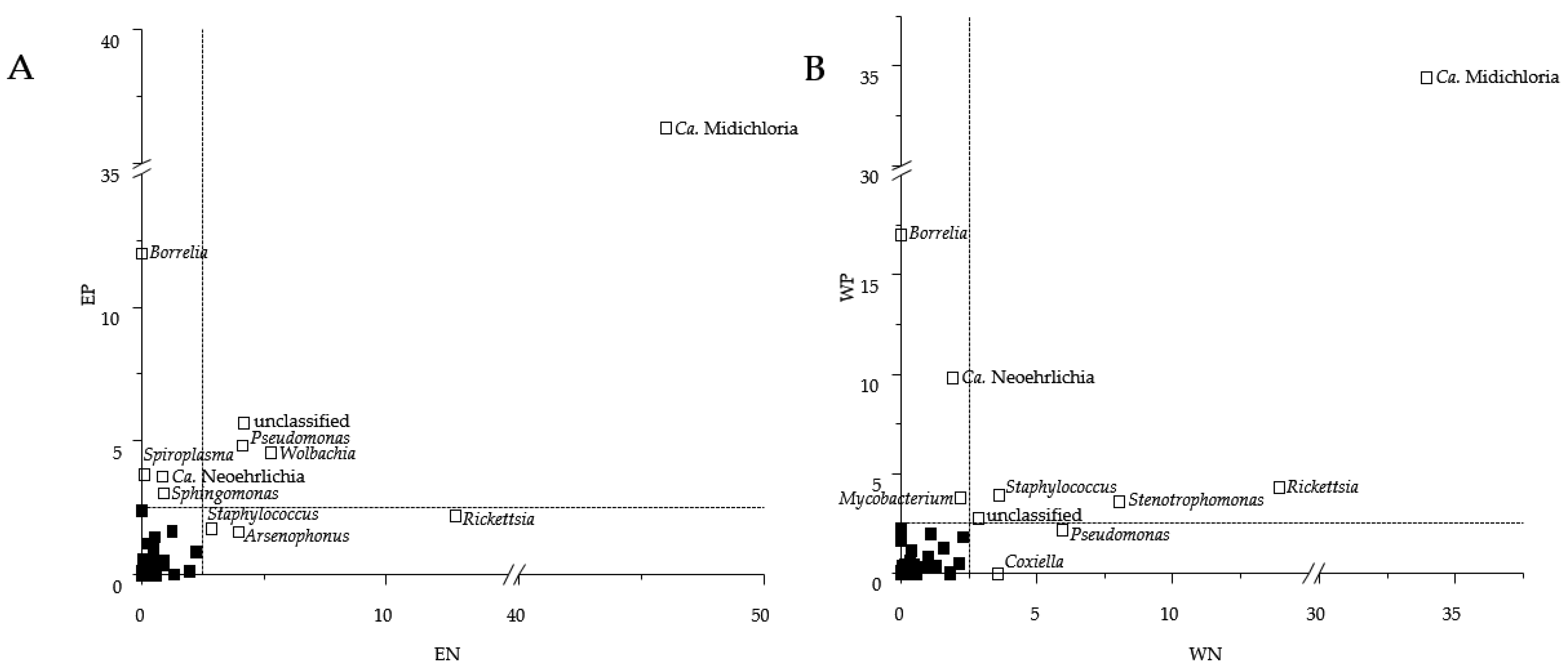
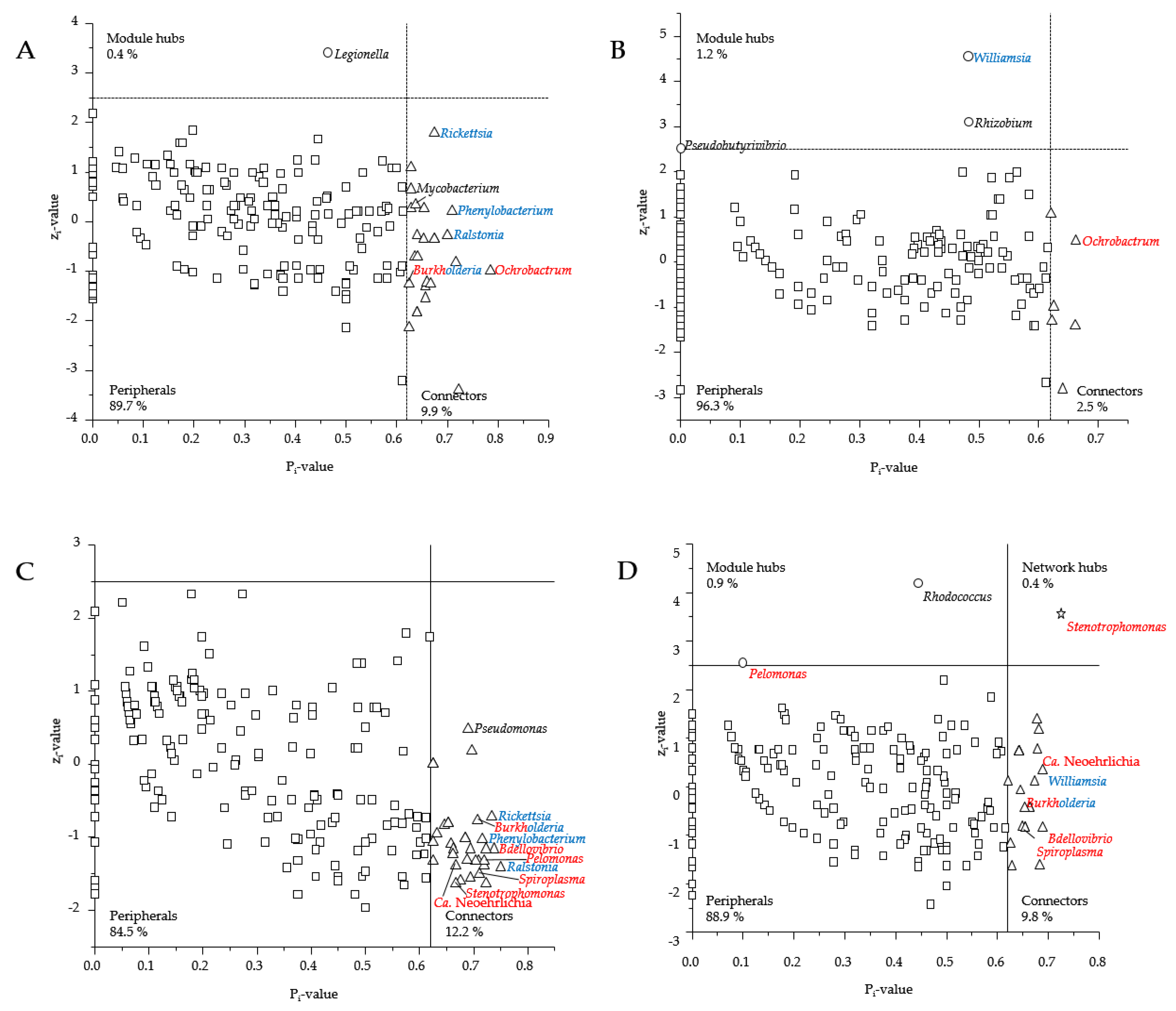
3.3. Presence of Human Pathogenic Bb Species Shifted Co-Occurrence in the Tick Microbiome
4. Discussion
4.1. Presence of Human Pathogenic Bb Species Shifted Sequence Read Abundances of Bacterial Genera instead of Bacterial Community Composition
4.2. Is the Location Important for the Composition of the Tick Microbiome?
4.3. How Does the Presence of Human Pathogenic Bb Species Shift the Bacterial Networks?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabezas-Cruz, A.; Pollet, T.; Estrada-Peña, A.; Allain, E.; Bonnet, S.I.; Moutailler, S. Handling the Microbial Complexity Associated to Ticks. In Ticks and Tick-Borne Pathogens; Abubakar, M., Perera, P.K., Eds.; IntechOpen: London, UK, 2019; pp. 1–36. [Google Scholar]
- Madison-Antenucci, S.; Kramer, L.D.; Gebhardt, L.L.; Kauffman, E. Emerging Tick-Borne Diseases. Clin. Microbiol. Rev. 2020, 33, e00083-18. [Google Scholar] [CrossRef] [PubMed]
- Brisson, D.; Drecktrah, D.; Eggers, C.H.; Samuels, D.S. Genetics of Borrelia burgdorferi. Annu. Rev. Genet. 2012, 46, 515–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SurvStat@RKI 2.0: Web-Based Query of Reporting Data in Accordance with Infection Protection Act (IfSG). Available online: https://survstat.rki.de (accessed on 13 November 2020).
- Zubriková, D.; Wittmann, M.; Hönig, V.; Švec, P.; Víchová, B.; Essbauer, S.; Dobler, G.; Grubhoffer, L.; Pfister, K. Prevalence of tick-borne encephalitis virus and Borrelia burgdorferi sensu lato in Ixodes ricinus ticks in Lower Bavaria and Upper Palatinate, Germany. Ticks Tick Borne Dis. 2020, 11, 101375. [Google Scholar] [CrossRef] [PubMed]
- Stanek, G.; Reiter, M. The expanding Lyme Borrelia complex-clinical significance of genomic species? Clin. Microbiol. Infect. 2011, 17, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivacic, L.; Reed, K.D.; Mitchell, P.D.; Ghebranious, N. A LightCycler TaqMan assay for detection of Borrelia burgdorferi sensu lato in clinical samples. Diagn. Microbiol. Infect. Dis. 2007, 57, 137–143. [Google Scholar] [CrossRef]
- Wills, M.K.B.; Kirby, A.M.; Lloyd, V.K. Detecting the Lyme Disease Spirochete, Borrelia burgdorferi, in Ticks Using Nested PCR. J. Vis. Exp. 2018, 132, e56471. [Google Scholar] [CrossRef] [Green Version]
- Santino, I.; Berlutti, F.; Pantanella, F.; Sessa, R.; Del Piano, M. Detection of Borrelia burgdorferi sensu lato DNA by PCR in serum of patients with clinical symptoms of Lyme borreliosis. FEMS Microbiol. Lett. 2008, 283, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Jarguín, A.; Díaz-Sánchez, S.; Villar, M.; de La Fuente, J. Integrated metatranscriptomics and metaproteomics for the characterization of bacterial microbiota in unfed Ixodes ricinus. Ticks Tick Borne Dis. 2018, 9, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-C.; Yang, Z.-N.; Lu, B.; Ma, X.-F.; Zhang, C.-X.; Xu, H.-J. The composition and transmission of microbiome in hard tick, Ixodes persulcatus, during blood meal. Ticks Tick Borne Dis. 2014, 5, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Kmeť, V.; Čaplová, Z. An update of the Ixodes ricinus microbiome. JMBFS 2019, 8, 1340–1342. [Google Scholar] [CrossRef]
- Okła, H.; Sosnowska, M.; Jasik, K.P.; Słodki, J.; Wojtyczka, R.D. Nonspecific Bacterial Flora Isolated from the Body Surface and Inside Ixodes ricinus Ticks. Pol. J. Microbiol. 2012, 61, 205–209. [Google Scholar] [CrossRef]
- Hoffmann, A.; Fingerle, V.; Noll, M. Analysis of Tick Surface Decontamination Methods. Microorganisms 2020, 8, 987. [Google Scholar] [CrossRef] [PubMed]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasit. Vectors 2018, 11, 12. [Google Scholar] [CrossRef]
- Chauhan, G.; McClure, J.; Hekman, J.; Marsh, P.W.; Bailey, J.A.; Daniels, R.F.; Genereux, D.P.; Karlsson, E.K. Combining Citizen Science and Genomics to Investigate Tick, Pathogen, and Commensal Microbiome at Single-Tick Resolution. Front. Genet. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chicana, B.; Couper, L.I.; Kwan, J.Y.; Tahiraj, E.; Swei, A. Comparative Microbiome Profiles of Sympatric Tick Species from the Far-Western United States. Insects 2019, 10, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landesman, W.J.; Mulder, K.; Fredericks, L.P.; Allan, B.F. Cross-kingdom analysis of nymphal-stage Ixodes scapularis microbial communities in relation to Borrelia burgdorferi infection and load. FEMS Microbiol. Ecol. 2019, 95, fiz167. [Google Scholar] [CrossRef] [PubMed]
- Couper, L.I.; Kwan, J.Y.; Ma, J.; Swei, A. Drivers and patterns of microbial community assembly in a Lyme disease vector. Ecol. Evol. 2019, 9, 7768–7779. [Google Scholar] [CrossRef] [Green Version]
- Pollet, T.; Sprong, H.; Lejal, E.; Krawczyk, A.I.; Moutailler, S.; Cosson, J.-F.; Vayssier-Taussat, M.; Estrada-Peña, A. The scale affects our view on the identification and distribution of microbial communities in ticks. Parasit. Vectors 2020, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.I.; Pollet, T. Update on the intricate tango between tick microbiomes and tick-borne pathogens. Parasite Immunol. 2020, 43, e12813. [Google Scholar] [CrossRef]
- Asghar, N.; Petersson, M.; Johansson, M.; Dinnetz, P. Local landscape effects on population dynamics of Ixodes ricinus. Geospat. Health 2016, 11, 487. [Google Scholar] [CrossRef] [Green Version]
- Vaculová, T.; Derdáková, M.; Špitalská, E.; Václav, R.; Chvostáč, M.; Rusňáková Tarageľová, V. Simultaneous Occurrence of Borrelia miyamotoi, Borrelia burgdorferi sensu lato, Anaplasma phagocytophilum and Rickettsia helvetica in Ixodes ricinus Ticks in Urban Foci in Bratislava, Slovakia. Acta Parasitol. 2019, 64, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Venczel, R.; Knoke, L.; Pavlovic, M.; Dzaferovic, E.; Vaculova, T.; Silaghi, C.; Overzier, E.; Konrad, R.; Kolenčík, S.; Derdakova, M.; et al. A novel duplex real-time PCR permits simultaneous detection and differentiation of Borrelia miyamotoi and Borrelia burgdorferi sensu lato. Infection 2016, 44, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.R. Ticks of Domestic Animals in Africa: A Guide to Identification of Species; Bioscience Reports: Edinburgh, UK, 2003; pp. 138–157. [Google Scholar]
- Pebesma, E. Simple Features for R: Standardized Support for Spatial Vector Data. R J. 2018, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- R Studio: Integrated Development for, R. Available online: http://www.rstudio.com (accessed on 31 December 2020).
- Tennekes, M. tmap: Thematic Maps in R. J. Stat. Softw. 2018, 84, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016; pp. 87–93. [Google Scholar]
- Wickham, H.; Grolemund, G. R for Data Science: Import, Tidy, Transform, Visualize, and Model Data, 1st ed.; O’Reilly: Sebastopol, CA, USA, 2017; pp. 352–387. [Google Scholar]
- Grüne Liste der Naturschutzgebiete in der Oberpfalz. Available online: https://www.lfu.bayern.de/natur/schutzgebiete/schutzgebietslisten/doc/nsg_oberpfalz.pdf (accessed on 4 February 2021).
- Schutzgebietsstatistik Kreise. Available online: https://udo.lubw.baden-wuerttemberg.de/public/pages/home/welcome.xhtml (accessed on 4 February 2021).
- Climate Data of Esslingen am Neckar (Germany). Available online: https://de.climate-data.org/europa/deutschland/baden-wuerttemberg/esslingen-am-neckar-6496/ (accessed on 5 February 2021).
- Climate Data of Weiden in der Oberpfalz (Germany). Available online: https://de.climate-data.org/europa/deutschland/bayern/weiden-i-d-opf-46468/ (accessed on 5 February 2021).
- Norte, A.C.; Margos, G.; Becker, N.S.; Albino Ramos, J.; Núncio, M.S.; Fingerle, V.; Araújo, P.M.; Adamík, P.; Alivizatos, H.; Barba, E.; et al. Host dispersal shapes the population structure of a tick-borne bacterial pathogen. Mol. Ecol. 2020, 29, 485–501. [Google Scholar] [CrossRef]
- Margos, G.; Jungnick, S.; Rieger, M.; Koloczek, J.; Sing, A.; Fingerle, V. Molekulare Typisierungsverfahren bei Borrelia. Gesundheitswesen 2015, 77, A84. [Google Scholar] [CrossRef]
- Margos, G.; Gatewood, A.G.; Aanensen, D.M.; Hanincová, K.; Terekhova, D.; Vollmer, S.A.; Cornet, M.; Piesman, J.; Donaghy, M.; Bormane, A.; et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 2008, 105, 8730–8735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Noll, M.; Matthies, D.; Frenzel, P.; Derakshani, M.; Liesack, W. Succession of bacterial community structure and diversity in a paddy soil oxygen gradient. Environ. Microbiol. 2005, 7, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Soft. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 13 October 2020).
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [Green Version]
- Kitsos, C.; Toulias, T. Hellinger Distance Between Generalized Normal Distributions. BJMCS 2017, 21, 1–16. [Google Scholar] [CrossRef]
- Brown, M.B. 400: A Method for Combining Non-Independent, One-Sided Tests of Significance. Biometrics 1975, 31, 987. [Google Scholar] [CrossRef]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.J. A measure of betweenness centrality based on random walks. Soc. Netw. 2005, 27, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabási, A.L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar] [CrossRef] [Green Version]
- Brinkerhoff, R.J.; Clark, C.; Ocasio, K.; Gauthier, D.T.; Hynes, W.L. Factors affecting the microbiome of Ixodes scapularis and Amblyomma americanum. PLoS ONE 2020, 15, e0232398. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Bartkova, S.; Lindestad, O.; Råberg, L. Co-infection with ‘Candidatus Neoehrlichia Mikurensis’ and Borrelia afzelii in Ixodes ricinus ticks in southern Sweden. Vector Borne Zoonotic Dis. 2013, 13, 438–442. [Google Scholar] [CrossRef]
- Obiegala, A.; Jeske, K.; Augustin, M.; Król, N.; Fischer, S.; Mertens-Scholz, K.; Imholt, C.; Suchomel, J.; Heroldova, M.; Tomaso, H.; et al. Highly prevalent bartonellae and other vector-borne pathogens in small mammal species from the Czech Republic and Germany. Parasit. Vectors 2019, 12, 332. [Google Scholar] [CrossRef] [Green Version]
- Springer, A.; Raulf, M.-K.; Fingerle, V.; Strube, C. Borrelia prevalence and species distribution in ticks removed from humans in Germany, 2013-2017. Ticks Tick Borne Dis. 2020, 11, 101363. [Google Scholar] [CrossRef]
- Rauter, C.; Hartung, T. Prevalence of Borrelia burgdorferi sensu lato genospecies in Ixodes ricinus ticks in Europe: A metaanalysis. Appl. Environ. Microbiol. 2005, 71, 7203–7216. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.S.; Goudreau, A.; Bissonnette, N.; Shamputa, I.C.; Tahlan, K. Methods for Detecting Mycobacterial Mixed Strain Infections-A Systematic Review. Front. Genet. 2020, 11, 600692. [Google Scholar] [CrossRef]
- Cabello, F.C.; Godfrey, H.P.; Bugrysheva, J.V.; Newman, S.A. Sleeper cells: The stringent response and persistence in the Borreliella (Borrelia) burgdorferi enzootic cycle. Environ. Microbiol. 2017, 19, 3846–3862. [Google Scholar] [CrossRef] [Green Version]
- Escobedo-Hinojosa, W.; Pardo-López, L. Analysis of bacterial metagenomes from the Southwestern Gulf of Mexico for pathogens detection. Pathog. Dis. 2017, 75, ftx058. [Google Scholar] [CrossRef]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Ansari, J.M.; Jefferson, K.K.; Cava, F.; Jacobs-Wagner, C.; et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.-J.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Kowalec, M.; Szewczyk, T.; Welc-Falęciak, R.; Siński, E.; Karbowiak, G.; Bajer, A. Rickettsiales Occurrence and Co-occurrence in Ixodes ricinus Ticks in Natural and Urban Areas. Microb. Ecol. 2019, 77, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Ross, B.D.; Hayes, B.; Radey, M.C.; Lee, X.; Josek, T.; Bjork, J.; Neitzel, D.; Paskewitz, S.; Chou, S.; Mougous, J.D. Ixodes scapularis does not harbor a stable midgut microbiome. ISME J. 2018, 12, 2596–2607. [Google Scholar] [CrossRef] [Green Version]
- Plantard, O.; Bouju-Albert, A.; Malard, M.-A.; Hermouet, A.; Capron, G.; Verheyden, H. Detection of Wolbachia in the tick Ixodes ricinus is due to the presence of the hymenoptera endoparasitoid Ixodiphagus hookeri. PLoS ONE 2012, 7, e30692. [Google Scholar] [CrossRef] [Green Version]
- Stafford, K.C.; Denicola, A.J.; Kilpatrick, H.J. Reduced abundance of Ixodes scapularis (Acari: Ixodidae) and the tick parasitoid Ixodiphagus hookeri (Hymenoptera: Encyrtidae) with reduction of white-tailed deer. J. Med. Entomol. 2003, 40, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Aivelo, T.; Norberg, A.; Tschirren, B. Bacterial microbiota composition of Ixodes ricinus ticks: The role of environmental variation, tick characteristics and microbial interactions. PeerJ 2019, 7, e8217. [Google Scholar] [CrossRef] [Green Version]
- Kwan, J.Y.; Griggs, R.; Chicana, B.; Miller, C.; Swei, A. Vertical vs. horizontal transmission of the microbiome in a key disease vector, Ixodes pacificus. Mol. Ecol. 2017, 26, 6578–6589. [Google Scholar] [CrossRef]
- Thapa, S.; Zhang, Y.; Allen, M.S. Effects of temperature on bacterial microbiome composition in Ixodes scapularis ticks. Microbiologyopen 2019, 8, e00719. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Peña, A.; Cabezas-Cruz, A.; Pollet, T.; Vayssier-Taussat, M.; Cosson, J.-F. High Throughput Sequencing and Network Analysis Disentangle the Microbial Communities of Ticks and Hosts Within and Between Ecosystems. Front. Cell. Infect. Microbiol. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.F.K. Milgram (1967): The Small World Problem. In Schlüsselwerke der Netzwerkforschung; Holzer, B., Stegbauer, C., Eds.; Springer: Wiesbaden, Germany, 2019; pp. 407–410. [Google Scholar]
- Lu, L.; Yin, S.; Liu, X.; Zhang, W.; Gu, T.; Shen, Q.; Qiu, H. Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The Tick Microbiome: Why Non-pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 1–14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EN | EP | WN | WP | |||||
|---|---|---|---|---|---|---|---|---|
| Interacting Bacterial Genera | Mutual Exclusions | Co-Occurence | Mutual Exclusions | Co-Occurence | Mutual Exclusions | Co-Occurence | Mutual Exclusions | Co-Occurence |
| Borrelia | / | / | / | 1 | / | / | 1 | 1 |
| Rickettsia | 106 | 0 | 2 | 0 | 25 | 3 | 8 | 0 |
| Mycobacterium | 13 | 36 | 14 | 5 | 8 | 29 | 1 | 11 |
| Phenylobacterium | 14 | 10 | 11 | 8 | 5 | 4 | 1 | 8 |
| Ralstonia | 5 | 5 | 15 | 5 | 12 | 11 | 2 | 18 |
| Burkholderia | 1 | 3 | 2 | 6 | 23 | 8 | 7 | 15 |
| Ochrobactrum | 6 | 12 | 56 | 6 | 1 | 23 | 7 | 3 |
| Legionella | 3 | 14 | 0 | 20 | 0 | 35 | 3 | 10 |
| Williamsia | 2 | 36 | 41 | 24 | 10 | 35 | 5 | 37 |
| Rhizobium | 0 | 37 | 31 | 19 | 2 | 13 | 20 | 8 |
| Pseudobutyrivibrio | / | / | 7 | 22 | 2 | 37 | 2 | 18 |
| Pseudomonas | 8 | 2 | 5 | 0 | 16 | 5 | 5 | 6 |
| Bdellovibrio | 1 | 6 | / | / | 7 | 11 | 10 | 16 |
| Pelomonas | 1 | 8 | 5 | 8 | 3 | 9 | 17 | 22 |
| Spiroplasma | 2 | 0 | 7 | 3 | 4 | 3 | 13 | 3 |
| Stenotrophomonas | 2 | 4 | 3 | 10 | 36 | 9 | 60 | 3 |
| Ca. Neoehrlichia | 2 | 1 | 5 | 4 | 9 | 2 | 25 | 6 |
| Rhodococcus | 0 | 32 | 4 | 27 | 4 | 22 | 30 | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Müller, T.; Fingerle, V.; Noll, M. Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations. Microorganisms 2021, 9, 1814. https://doi.org/10.3390/microorganisms9091814
Hoffmann A, Müller T, Fingerle V, Noll M. Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations. Microorganisms. 2021; 9(9):1814. https://doi.org/10.3390/microorganisms9091814
Chicago/Turabian StyleHoffmann, Angeline, Thomas Müller, Volker Fingerle, and Matthias Noll. 2021. "Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations" Microorganisms 9, no. 9: 1814. https://doi.org/10.3390/microorganisms9091814
APA StyleHoffmann, A., Müller, T., Fingerle, V., & Noll, M. (2021). Presence of Human Pathogens of the Borrelia burgdorferi sensu lato Complex Shifts the Sequence Read Abundances of Tick Microbiomes in Two German Locations. Microorganisms, 9(9), 1814. https://doi.org/10.3390/microorganisms9091814