Novel Genomic Regions Associated with Intramuscular Fatty Acid Composition in Rabbits

 ,
,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Phenotypes
2.3. Genotypes and Quality Control
2.4. Genome-Wide Association Study (GWAS)
2.5. Genomic Regions Associated with the Fatty Acid Composition
2.6. Identification of Candidate Genes
2.7. Linkage Disequilibrium Analysis
2.8. Internal Validation
2.8.1. Permutation Test
2.8.2. Within line GWAS
2.9. Ethics Statement
3. Results and Discussion
3.1. Descriptive Statistics of the Intramuscular Fatty Acid Composition
3.2. Genomic Data
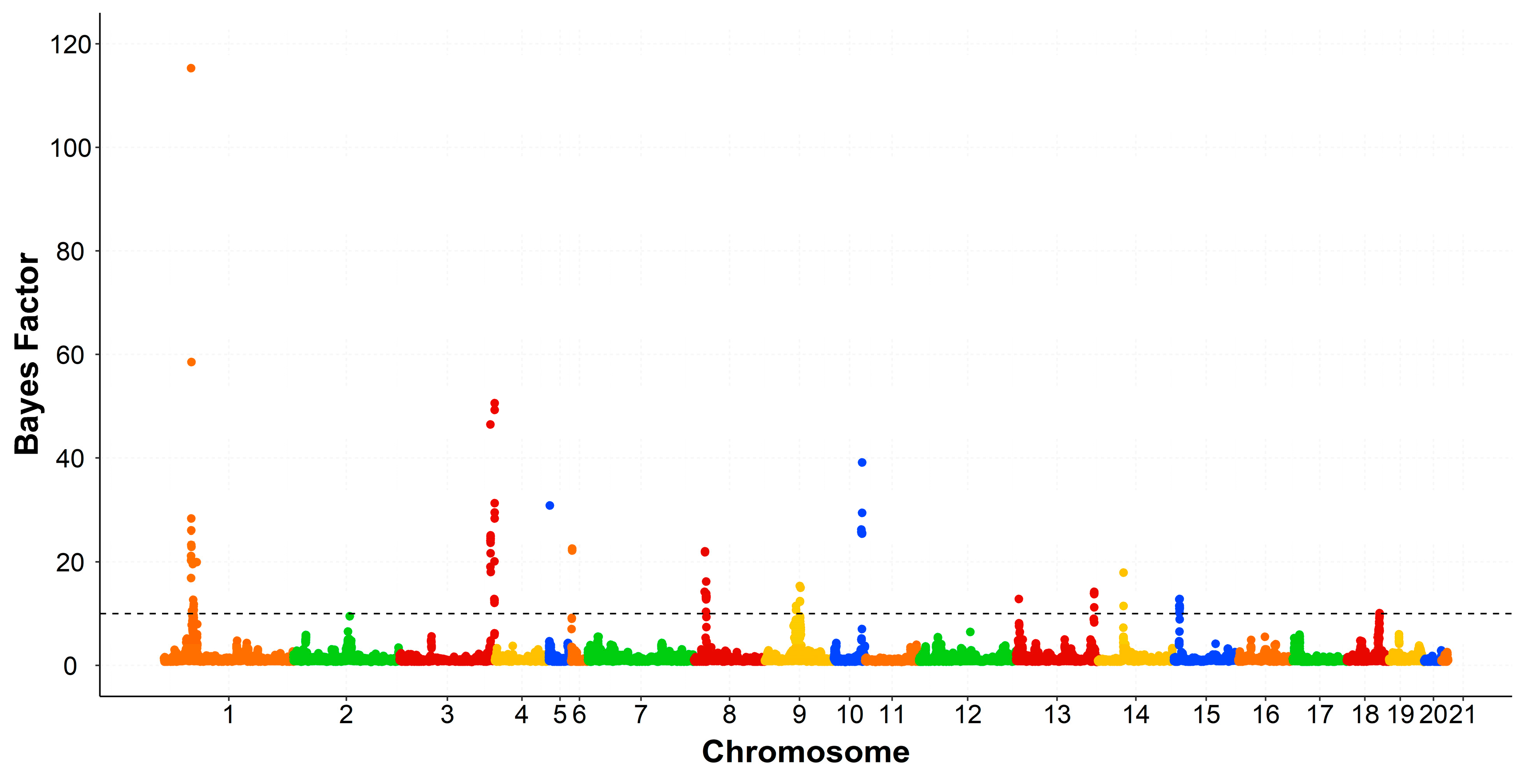
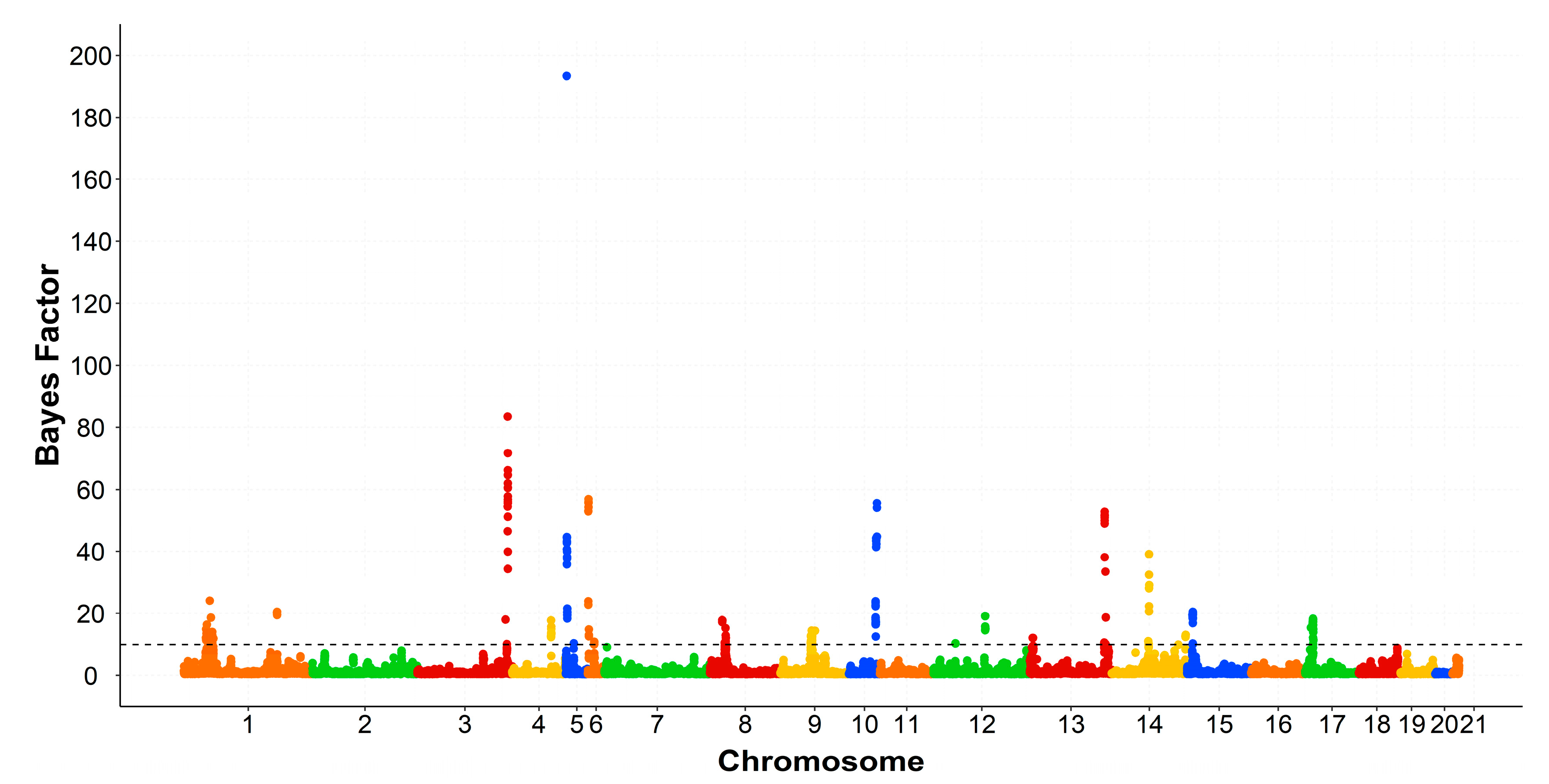
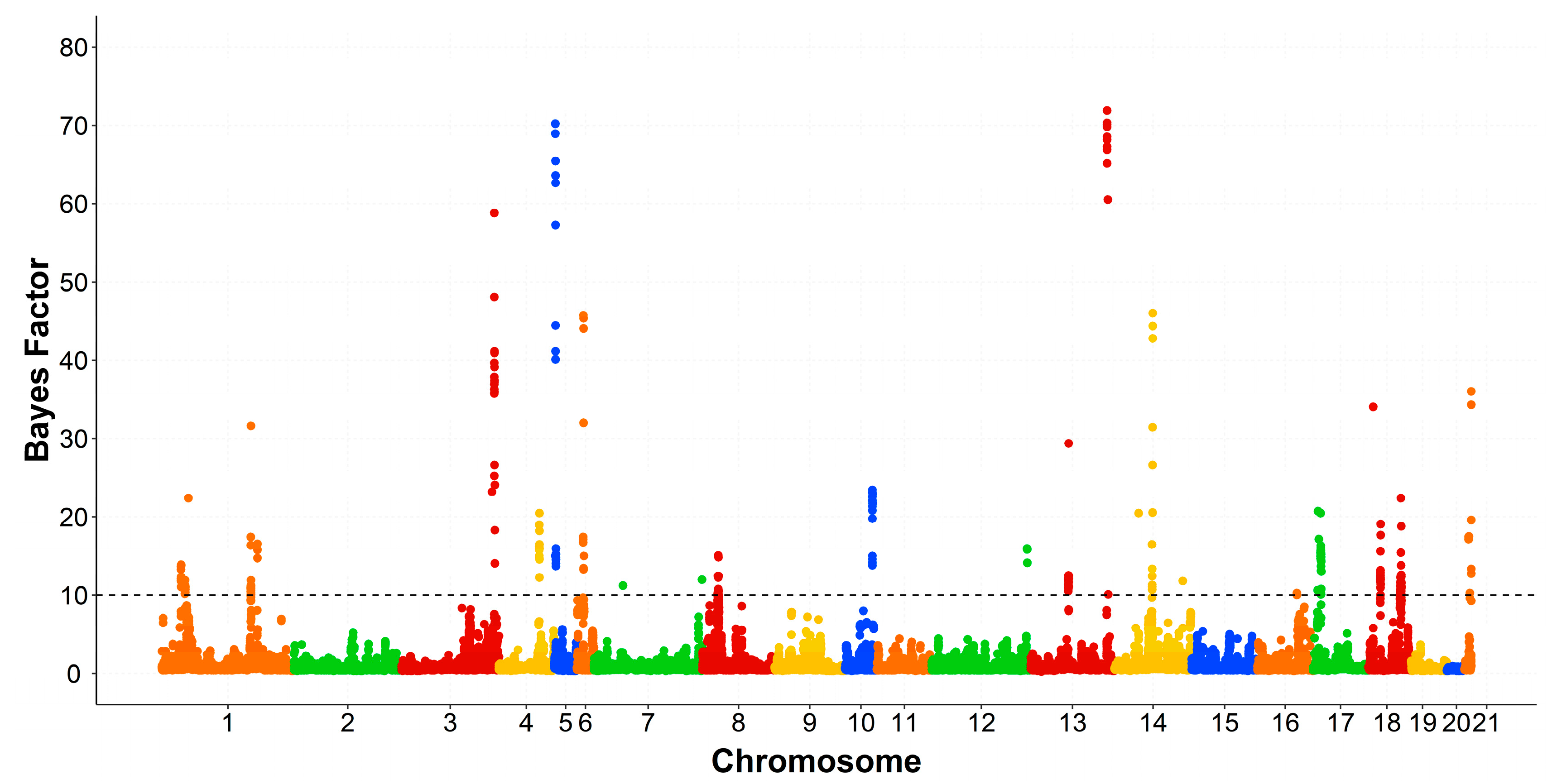
3.3. Genomic Regions Associated with the Intramuscular Fatty Acid Composition
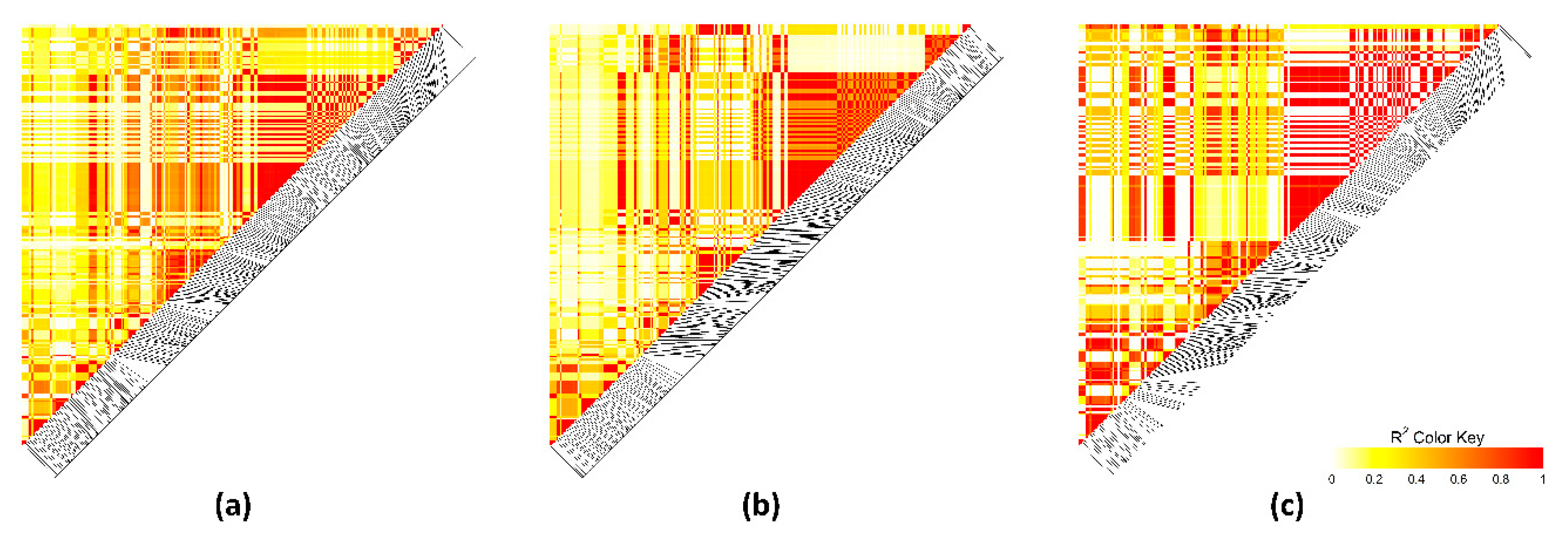
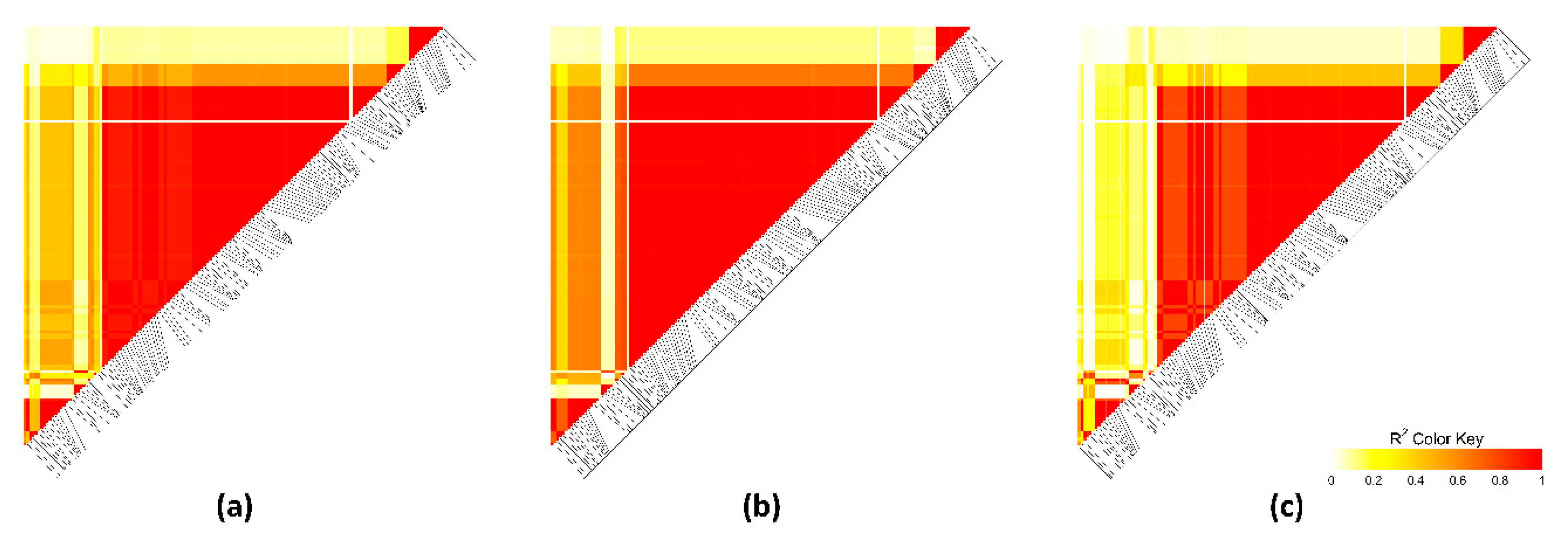
3.4. Linkage Disequilibrium Analysis
3.5. Polygenic Nature of the Intramuscular Fatty Acid Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hernández, P.; Zotte, A.D. Influence of diet on Rabbit meat quality. In Nutrition of the Rabbit; CABI Publishing: Wallingford, UK, 2020; pp. 172–192. [Google Scholar]
- Martins, T.D.S.; De Lemos, M.V.A.; Mueller, L.F.; Baldi, F.; Amorim, T.; Ferrinho, A.M.; Muñoz, J.A.; Fuzikawa, I.H.D.S.; De Moura, G.V.; Gemelli, J.L.; et al. Fat Deposition, Fatty Acid Composition, and Its Relationship with Meat Quality and Human Health. Meat Sci. Nutr. 2018, 18–37. [Google Scholar] [CrossRef] [Green Version]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar] [CrossRef]
- Nettleton, J.A.; Brouwer, I.A.; Geleijnse, J.M.; Hornstra, G. Saturated Fat Consumption and Risk of Coronary Heart Disease and Ischemic Stroke: A Science Update. Ann. Nutr. Metab. 2017, 70, 26–33. [Google Scholar] [CrossRef]
- Sapp, R.L.; Bertrand, J.K.; Pringle, T.D.; Wilson, D.E. Effects of selection for ultrasound intramuscular fat percentage in Angus bulls on carcass traits of progeny. J. Anim. Sci. 2002, 80, 2017–2022. [Google Scholar] [CrossRef]
- Liu, L.; Cui, H.; Xing, S.; Zhao, G.; Wen, J. Effect of Divergent Selection for Intramuscular Fat Content on Muscle Lipid Metabolism in Chickens. Anim. 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.P.; Chen, J.L.; Zheng, M.Q.; Wen, J.; Zhang, Y. Correlated Responses to Selection for Increased Intramuscular Fat in a Chinese Quality Chicken Line. Poult. Sci. 2007, 86, 2309–2314. [Google Scholar] [CrossRef]
- Schwab, C.R.; Baas, T.J.; Stalder, K.J.; Nettleton, D. Results from six generations of selection for intramuscular fat in Duroc swine using real-time ultrasound. I. Direct and correlated phenotypic responses to selection1. J. Anim. Sci. 2009, 87, 2774–2780. [Google Scholar] [CrossRef]
- Martínez-Álvaro, M.; Blasco, A.; Hernández, P. Effect of selection for intramuscular fat on the fatty acid composition of rabbit meat. Animal 2018, 12, 2002–2008. [Google Scholar] [CrossRef]
- Sosa-Madrid, B.S.; Hernández, P.; Blasco, A.; Haley, C.S.; Fontanesi, L.; Santacreu, M.A.; Pena, R.N.; Navarro, P.; Ibáñez-Escriche, N. Genomic regions influencing intramuscular fat in divergently selected rabbit lines. Anim. Genet. 2019, 51, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Gol, S.; Pena, R.N.; Tor, M.; Ibáñez-Escriche, N.; Dekkers, J.C.M.; Estany, J. Genome-Wide Association Study Singles Out SCD and LEPR as the Two Main Loci Influencing Intramuscular Fat Content and Fatty Acid Composition in Duroc Pigs. PLoS ONE 2016, 11, e0152496. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, J.; Cui, L.; Ma, J.; Chen, C.; Ai, H.; Xie, X.; Li, L.; Xiao, S.; Huang, L.; et al. Genetic architecture of fatty acid composition in the longissimus dorsi muscle revealed by genome-wide association studies on diverse pig populations. Genet. Sel. Evol. 2016, 48, 5. [Google Scholar] [CrossRef] [Green Version]
- Pena, R.N.; Noguera, J.L.; García-Santana, M.J.; González, E.; Tejeda, J.F.; Ros-Freixedes, R.; Ibáñez-Escriche, N. Five genomic regions have a major impact on fat composition in Iberian pigs. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cesar, A.S.M.; Coutinho, L.L.; Regitano, L.C.A.; Mourão, G.B.; Tullio, R.R.; Lanna, D.P.D.; Nassu, R.T.; Mudado, M.A.; De Oliveira, P.S.N.; Nascimento, M.L.D.; et al. Genome-wide association study for intramuscular fat deposition and composition in Nellore cattle. BMC Genet. 2014, 15, 39. [Google Scholar] [CrossRef] [Green Version]
- Sasago, N.; Abe, T.; Sakuma, H.; Kojima, T.; Uemoto, Y. Genome-wide association study for carcass traits, fatty acid composition, chemical composition, sugar, and the effects of related candidate genes in Japanese Black cattle. Anim. Sci. J. 2016, 88, 33–44. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, B.; Niu, H.; Zhang, W.; Xu, L.; Xu, L.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; et al. Genome wide association study identifies SNPs associated with fatty acid composition in Chinese Wagyu cattle. J. Anim. Sci. Biotechnol. 2019, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Zomeño, C.; Hernández, P.; Blasco, A. Divergent selection for intramuscular fat content in rabbits. I. Direct response to selection1. J. Anim. Sci. 2013, 91, 4526–4531. [Google Scholar] [CrossRef]
- Zomeño, C.; Juste, V.; Hernández, P. Application of NIRS for predicting fatty acids in intramuscular fat of rabbit. Meat Sci. 2012, 91, 155–159. [Google Scholar] [CrossRef]
- O’Fallon, J.V.; Busboom, J.R.; Nelson, M.L.; Gaskins, C.T. A direct method for fatty acid methyl ester synthesis: Application to wet meat tissues, oils, and feedstuffs. J. Anim. Sci. 2007, 85, 1511–1521. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Blasco, A. Bayesian Data Analysis for Animal Scientists; Springer Science and Business Media LLC: New York, NY, USA, 2017. [Google Scholar]
- Garrick, D.J.; Fernando, R.L. Implementing a QTL Detection Study (GWAS) Using Genomic Prediction Methodology. Advanced Structural Safety Studies 2013, 1019, 275–298. [Google Scholar]
- Bouwman, A.C.; Janss, L.L.G.; Heuven, H. A Bayesian approach to detect QTL affecting a simulated binary and quantitative trait. BMC Proc. 2011, 5, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773. [Google Scholar] [CrossRef]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.E.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2018, 47, D745–D751. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.M.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Shin, J.-H.; Blay, S.; Graham, J.; McNeney, B. LDheatmap: An R Function for Graphical Display of Pairwise Linkage Disequilibria Between Single Nucleotide Polymorphisms. J. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Casto-Rebollo, C.; Argente, M.J.; García, M.L.; Pena, R.; Ibáñez-Escriche, N. Identification of functional mutations associated with environmental variance of litter size in rabbits. Genet. Sel. Evol. 2020, 52, 1–9. [Google Scholar] [CrossRef]
- Kessner, D.; Novembre, J. Power Analysis of Artificial Selection Experiments Using Efficient Whole Genome Simulation of Quantitative Traits. Genet. 2015, 199, 991–1005. [Google Scholar] [CrossRef]
- Lou, R.N.; Therkildsen, N.O.; Messer, P.W. The Effects of Quantitative Trait Architecture on Detection Power in Short-Term Artificial Selection Experiments. G3 Genes Genomes Genet. 2020, 10, 3213–3227. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Jin, J.; Hu, W.; Sun, W.; Bielawski, J.; Szulc, Z.; Taha, T.; Obeid, L.M.; Mao, C. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. 2006, 20, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zou, F.; Diao, Z.; Zhang, S.; Deng, Y.; Zhu, X.; Cui, L.; Yu, J.; Zhang, Z.; Bamigbade, A.T.; et al. Perilipin 2 and lipid droplets provide reciprocal stabilization. Biophys. Rep. 2019, 5, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, G.; Mazzoni, M.; Zambonelli, P.; Lalatta-Costerbosa, G.; Tronca, A.; Russo, V.; Davoli, R. Perilipin 1 and perilipin 2 protein localization and gene expression study in skeletal muscles of European cross-breed pigs with different intramuscular fat contents. Meat Sci. 2011, 88, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Gol, S.; Ros-Freixedes, R.; Zambonelli, P.; Tor, M.; Pena, R.N.; Braglia, S.; Zappaterra, M.; Estany, J.; Davoli, R. Relationship between perilipin genes polymorphisms and growth, carcass and meat quality traits in pigs. J. Anim. Breed. Genet. 2015, 133, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Sosa-Madrid, B.S.; Varona, L.; Blasco, A.; Hernández, P.; Casto-Rebollo, C.; Ibáñez-Escriche, N. The effect of divergent selection for intramuscular fat on the domestic rabbit genome. Animal 2020, 14, 2225–2235. [Google Scholar] [CrossRef]
- Kim, E.-S.; Ros-Freixedes, R.; Pena, R.N.; Baas, T.J.; Estany, J.; Rothschild, M.F. Identification of signatures of selection for intramuscular fat and backfat thickness in two Duroc populations1. J. Anim. Sci. 2015, 93, 3292–3302. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Lin, S.; Mu, H.; Tang, X.; Ou, Y.; Chen, J.; Ma, Y.; Li, Y. Analysis of Differentially Expressed Genes and Signaling Pathways Related to Intramuscular Fat Deposition in Skeletal Muscle of Sex-Linked Dwarf Chickens. BioMed. Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Ballester, M.; Ramayo-Caldas, Y.; Revilla, M.; Corominas, J.; Castelló, A.; Estellé, J.; Fernández, A.I.; Folch, J.M. Integration of liver gene co-expression networks and eGWAs analyses highlighted candidate regulators implicated in lipid metabolism in pigs. Sci. Rep. 2017, 7, 46539. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Liu, H.; Li, X.; Wei, W.; Zhao, S.; Cao, J. A genome-wide association study of five meat quality traits in Yorkshire pigs. Front. Agric. Sci. Eng. 2014, 1, 137–143. [Google Scholar] [CrossRef]
- Aung, H.W.; Henry, S.A.; Walker, L.P. Revising the Representation of Fatty Acid, Glycerolipid, and Glycerophospholipid Metabolism in the Consensus Model of Yeast Metabolism. Ind. Biotechnol. 2013, 9, 215–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janani, C.; Kumari, B.D.R. PPAR gamma gene A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Kongsuwan, K.; Greenwood, P.L.; Zhou, G.; Zhang, W.; Dalrymple, B.P. A gene expression estimator of intramuscular fat percentage for use in both cattle and sheep. J. Anim. Sci. Biotechnol. 2014, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xiong, K.; Sun, W.; Fu, Y.; Jiang, Z.; Yu, D.; Liu, H.; Chen, J. Two completely linked polymorphisms in thePPARGtranscriptional regulatory region significantly affect gene expression and intramuscular fat deposition in the longissimus dorsi muscle of Erhualian pigs. Anim. Genet. 2013, 44, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Kim, J.S.; Nguyen, T.H.; Lee, H.-J.; Baik, M. Wnt/β-catenin signaling and adipogenic genes are associated with intramuscular fat content in the longissimus dorsi muscle of Korean cattle. Anim. Genet. 2013, 44, 627–635. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.K.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nat. Cell Biol. 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Karbowska, J.; Kochan, Z. Role of adiponectin in the regulation of carbohydrate and lipid metabolism. J. Physiol. Pharmacol. 2006, 57, 103–113. [Google Scholar]
- Barendse, W. Haplotype Analysis Improved Evidence for Candidate Genes for Intramuscular Fat Percentage from a Genome Wide Association Study of Cattle. PLoS ONE 2011, 6, e29601. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Bower, N.I.; Reverter, A.; Tan, S.H.; De Jager, N.; Wang, R.; McWilliam, S.M.; Cafe, L.M.; Greenwood, P.L.; Lehnert, S.A. Gene expression patterns during intramuscular fat development in cattle1. J. Anim. Sci. 2009, 87, 119–130. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Y.; Mukiibi, R.; Chen, L.; Vinsky, M.; Plastow, G.S.; Basarab, J.A.; Stothard, P.; Li, C. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: Feed efficiency and component traits. BMC Genom. 2020, 21, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Maharani, D.; Park, H.-B.; Lee, J.-B.; Yoo, C.-K.; Lim, H.-T.; Han, S.-H.; Lee, S.-S.; Ko, M.-S.; Cho, I.-C.; Lee, J.H. Association of the gene encoding stearoyl-CoA desaturase (SCD) with fatty acid composition in an intercross population between Landrace and Korean native pigs. Mol. Biol. Rep. 2012, 40, 73–80. [Google Scholar] [CrossRef]
- Ramírez, C.A.; Horcada, A.; Polvillo, O.; Membrillo, A.; Anaya, G.; Molina, A.; Alcalde, M.J.; Panea, B. Association study between variability in the SCD gene and the fatty acid profile in perirenal and intramuscular fat deposits from Spanish goat populations. Small Rumin. Res. 2016, 136, 127–131. [Google Scholar] [CrossRef]
- Puig-Oliveras, A.; Revilla, M.; Castelló, A.; Fernández, A.I.; Folch, J.M.; Ballester, M. Expression-based GWAS identifies variants, gene interactions and key regulators affecting intramuscular fatty acid content and composition in porcine meat. Sci. Rep. 2016, 6, 31803. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | SD 1 | CV 2 (%) |
|---|---|---|---|
| C14:0 | 1.26 | 0.26 | 20 |
| C16:0 | 25.7 | 1.17 | 4 |
| C18:0 | 8.34 | 0.66 | 8 |
| SFA | 36.4 | 0.94 | 2 |
| C16:1n7 | 1.25 | 0.50 | 39 |
| C18:1n9 | 20.3 | 0.88 | 4 |
| MUFA | 24.4 | 1.27 | 5 |
| C18:2n6 | 27.7 | 1.18 | 4 |
| C18:3n3 | 1.16 | 0.14 | 12 |
| C20:4n6 | 7.45 | 1.05 | 14 |
| PUFA | 39.2 | 1.83 | 4 |
| PUFA/SFA | 1.08 | 0.07 | 6 |
| MUFA/SFA | 0.67 | 0.03 | 4 |
| n-6/n-3 | 17.7 | 0.97 | 5 |
| OCU 1 | Region (Mb) 2 | Trait | VAR 3 | PPA 4 | rSNP 5 | Number of Genes 6 | PCG 7 |
|---|---|---|---|---|---|---|---|
| 1 | 29.1–29.9 | C18:0 | 0.52 | 0.55 | 21 | 3 | - |
| 34.0–37.9 | C14:0 | 3.54 | 0.51 | 38 | 13 | ACER2, PLIN2 | |
| C16:0 | 11.20 | 0.51 | 36 | ||||
| C18:0 | 0.87 | 0.33 | 1 | ||||
| SFA | 11.32 | 0.45 | 22 | ||||
| 121.0–121.9 | C14:0 | 0.62 | 0.69 | 27 | 13 | MTMR2 | |
| C18:0 | 0.50 | 0.59 | 27 | ||||
| 3 | 149.0–149.9 | C14:0 | 1.11 | 0.96 | 19 | 8 | ENSOCUG00000000157 |
| C16:0 | 3.65 | 0.98 | 15 | ||||
| C18:0 | 1.72 | 0.99 | 15 | ||||
| SFA | 0.98 | 0.51 | 16 | ||||
| 154.0–154.9 | SFA | 0.91 | 0.39 | 12 | 4 | - | |
| 4 | 67.0–67.9 | C18:0 | 0.71 | 0.75 | 14 | 8 | - |
| 5 | 7.2–7.9 | C16:0 | 0.71 | 0.50 | 15 | 1 | - |
| 6 | 0–0.9 | C16:0 | 1.33 | 0.60 | 12 | - | - |
| 8 | 15.0–15.9 | SFA | 0.52 | 0.43 | 25 | 7 | - |
| 21.0–21.9 | C18:0 | 0.57 | 0.75 | 69 | 6 | PIK3C2G, PLCZ1, PLEKHA5 | |
| 9 | 64.0–64.9 | C16:0 | 0.62 | 0.38 | 2 | 10 | NPC1 |
| SFA | 0.96 | 0.37 | 2 | ||||
| 69.0–69.5 | SFA | 0.56 | 0.32 | 2 | 4 | - | |
| 10 | 43.0–43.9 | SFA | 0.59 | 0.35 | 8 | 3 | - |
| 13 | 126.0–126.9 | C18:0 | 0.96 | 0.87 | 9 | 23 | HEYL, MFSD2A, PPT1 |
| 14 | 82.0–82.9 | C18:0 | 0.51 | 0.55 | 18 | 19 | ADIPOQ |
| 15 | 10.0–10.9 | C16:0 | 0.63 | 0.40 | 17 | 11 | LRAT, TLR2 |
| SFA | 0.63 | 0.32 | 16 | ||||
| 18 | 48.0–48.9 | C18:0 | 0.52 | 0.52 | 42 | 32 | ERLIN1, |
| SFA | 0.55 | 0.41 | 2 | ENSOCUG00000014801 ENSOCUG00000001375 |
| OCU 1 | Region (Mb) 2 | Trait | VAR 3 | PPA 4 | rSNP 5 | Number of Genes 6 | PCG 7 |
|---|---|---|---|---|---|---|---|
| 1 | 29.1–29.9 | C16:1n7 | 0.81 | 0.48 | 16 | 3 | - |
| 36.0–36.9 | C16:1n7 | 0.92 | 0.34 | 3 | 2 | - | |
| 121.0–121.9 | C18:1n9 | 0.63 | 0.34 | 2 | 13 | MTMR2 | |
| MUFA | 0.61 | 0.38 | 18 | ||||
| 3 | 149.0–149.9 | C18:1n9 | 1.06 | 0.64 | 17 | 11 | ENSOCUG00000000157 |
| MUFA | 1.38 | 0.77 | 18 | ||||
| 4 | 67.0–67.9 | C18:1n9 | 0.66 | 0.41 | 14 | 8 | - |
| 5 | 7.2–7.9 | C16:1n7 | 0.82 | 0.61 | 22 | 1 | - |
| MUFA | 0.99 | 0.64 | 22 | ||||
| 6 | 8–9.9 | C16:1n7 | 0.71 | 0.64 | 22 | 22 | SMG1, GDE1 |
| C18:1n9 | 1.43 | 0.50 | 12 | ||||
| MUFA | 0.67 | 0.47 | 10 | ||||
| 8 | 21.0–21.9 | C16:1n7 | 0.63 | 0.65 | 8 | 6 | PIK3C2G, PLCZ1, |
| C18:1n9 | 0.72 | 0.57 | 4 | PLEKHA5 | |||
| MUFA | 1.01 | 0.66 | 16 | ||||
| 9 | 12.0–12.5 | C16:1n7 | 1.05 | 0.60 | 17 | 7 | PPARG |
| 10 | 42.2–42.9 | MUFA | 0.53 | 0.41 | 16 | 1 | - |
| 13 | 126.0–126.9 | C18:1n9 | 1.28 | 0.73 | 9 | 23 | HEYL, MFSD2A, PPT1 |
| MUFA | 0.99 | 0.69 | 9 | ||||
| 14 | 82.0–82.9 | C18:1n9 | 0.58 | 0.30 | 7 | 14 | ADIPOQ |
| 17 | 12.0–13.9 | C16:1n7 | 1.55 | 0.66 | 35 | 14 | ALDH1A2, LIPC, MYO1E |
| MUFA | 0.81 | 0.56 | 36 | ||||
| 18 | 47.0–48.9 | C16:1n7 | 1.21 | 0.43 | 7 | 35 | GOT1, ERLIN1, |
| C18:1n9 | 0.83 | 0.37 | 2 | ENSOCUG00000014801, | |||
| MUFA | 1.90 | 0.53 | 46 | ENSOCUG00000001375 | |||
| 21 | 10.0–10.9 | C18:1n9 | 0.72 | 0.44 | 6 | 2 | - |
| OCU 1 | Region (Mb) 2 | Trait | Var 3 | PPA 4 | rSNPs 5 | Number of Genes 6 | PCG 7 |
|---|---|---|---|---|---|---|---|
| 1 | 29.1–29.9 | PUFA | 0.56 | 0.34 | 15 | 3 | - |
| 31–31.9 | C18:2n6 | 1.34 | 0.27 | 2 | 7 | - | |
| PUFA | 0.6 | 0.31 | 16 | ||||
| 34–37.9 | C18:2n6 | 3.18 | 0.20 | 2 | 13 | ACER2, PLIN2 | |
| C20:4n6 | 1.57 | 0.47 | 33 | ||||
| PUFA | 2.41 | 0.37 | 11 | ||||
| 2 | 85.0–85.9 | C18:2n6 | 1.24 | 0.24 | 8 | 3 | - |
| 3 | 25.0–26.9 | C18:2n6 | 6.05 | 0.65 | 41 | 6 | FGF1, NR3C1 |
| 149.0–149.9 | C18:2n6 | 0.68 | 0.24 | 1 | 8 | ENSOCUG00000000157 | |
| C20:4n6 | 0.82 | 0.92 | 16 | ||||
| PUFA | 2.25 | 0.97 | 15 | ||||
| 5 | 7.2–7.9 | PUFA | 0.91 | 0.66 | 22 | 1 | - |
| 6 | 0–0.9 | PUFA | 0.55 | 0.57 | 14 | - | - |
| 5.0–5.9 | C18:2n6 | 0.61 | 0.21 | 8 | 15 | LITAF, SNX29 | |
| 8 | 21.0–21.9 | PUFA | 0.78 | 0.67 | 14 | 5 | PIK3C2G, PLCZ1 |
| 50.0–50.9 | C18:2n6 | 1.06 | 0.30 | - | 6 | - | |
| 55.3–55.9 | C18:2n6 | 4.77 | 0.30 | 11 | 1 | - | |
| C18:3n3 | 0.99 | 0.38 | 18 | ||||
| 9 | 63.0–63.9 | C18:2n6 | 0.61 | 0.23 | 1 | 4 | - |
| 64.0–65.9 | PUFA | 1.9 | 0.46 | 30 | 16 | NPC1, OSBPL1A | |
| 115.0–115.9 | C18:2n6 | 1.21 | 0.38 | 16 | 3 | - | |
| 10 | 43.0–43.9 | PUFA | 0.58 | 0.47 | 17 | 3 | - |
| 12 | 7.0–7.9 | C20:4n6 | 1.48 | 0.93 | 10 | 8 | - |
| 13 | 33.0–34.9 | C18:2n6 | 3.42 | 0.49 | 13 | 60 | CRP, DCAF8, FCER1A, PIGM, ATP1A2 |
| 14 | 156.2–156.9 | C18:3n3 | 0.73 | 0.74 | 17 | 3 | - |
| 15 | 10.0–10.9 | C18:2n6 | 2.41 | 0.39 | 16 | 11 | LRAT, TLR2 |
| PUFA | 0.87 | 0.53 | 19 | ||||
| 70.0–71.9 | C18:2n6 | 3.59 | 0.50 | 4 | 12 | ANXA3, GK2 | |
| 16 | 62.0–62.9 | C18:2n6 | 0.59 | 0.23 | 6 | 9 | HSD11B1 |
| 71.0–72.9 | C18:3n3 | 1.67 | 0.43 | 10 | 7 | ENSOCUG00000006240 | |
| 17 | 9.0–10.9 | C18:2n6 | 3.87 | 0.41 | 21 | 10 | |
| 12.0–13.9 | C18:2n6 | 3.55 | 0.46 | 34 | 14 | ALDH1A2, LIPC, | |
| PUFA | 0.84 | 0.61 | MYO1E | ||||
| 18 | 23.0–23.8 | C18:2n6 | 1.18 | 0.36 | 17 | 8 | NRBF2 |
| 19 | 21.0–21.9 | C18:2n6 | 0.52 | 0.21 | 2 | 15 | ADAP2 |
| 21 | 10.0–10.9 | C18:3n3 | 0.55 | 0.68 | 6 | 2 | - |
| OCU 1 | Region (Mb) 2 | Trait | Var 3 | PPA 4 | rSNPs 5 | Number of Genes 6 | PCG 7 |
|---|---|---|---|---|---|---|---|
| 1 | 29.1–29.9 | PUFA/SFA | 0.53 | 0.32 | 14 | 3 | - |
| 31.1–31.9 | PUFA/SFA | 0.59 | 0.30 | 11 | 7 | - | |
| 34.0–37.9 | PUFA/SFA | 2.98 | 0.38 | 10 | 13 | ACER2, PLIN2 | |
| n-6/n-3 | 1.45 | 0.29 | 17 | ||||
| 3 | 118.0–118.9 | MUFA/SFA | 0.58 | 0.36 | 1 | 11 | - |
| 149.0–149.9 | PUFA/SFA | 2.45 | 0.98 | 15 | 8 | ENSOCUG00000000157 | |
| 5 | 7.3–7.9 | PUFA/SFA | 0.84 | 0.61 | 22 | 1 | - |
| 6 | 0.0–0.9 | PUFA/SFA | 1.09 | 0.74 | 14 | - | - |
| 8 | 51.0–51.9 | n-6/n-3 | 0.67 | 0.42 | 3 | 7 | - |
| 53.0–53.8 | MUFA/SFA | 0.53 | 0.28 | 11 | 14 | DGKH | |
| 9 | 64.0–65.9 | PUFA/SFA | 2.44 | 0.50 | 38 | 16 | NPC1, OSBPL1A |
| 10 | 5.0–5.6 | MUFA/SFA | 0.63 | 0.37 | 19 | 4 | ITGB8 |
| 43.0–43.9 | PUFA/SFA | 0.52 | 0.45 | 8 | 3 | - | |
| 14 | 78.2–78.9 | MUFA/SFA | 0.52 | 0.23 | 5 | 14 | ATP11B |
| 15 | 8.0–8.9 | n-6/n-3 | 0.83 | 0.48 | 30 | 13 | - |
| 10.0–10.9 | PUFA/SFA | 1.60 | 0.71 | 18 | 11 | LRAT, TLR2 | |
| 16 | 62.0–62.9 | MUFA/SFA | 0.59 | 0.17 | 3 | 9 | HSD11B1 |
| 71.0–72.9 | n-6/n-3 | 1.04 | 0.26 | 10 | 7 | ENSOCUG00000006240 | |
| 18 | 11.0–11.9 | MUFA/SFA | 0.86 | 0.32 | 3 | 5 | SAMD8 |
| 21.0–21.9 | MUFA/SFA | 0.93 | 0.52 | 22 | 3 | - | |
| 46.0–48.9 | MUFA/SFA | 7.91 | 0.59 | 54 | 41 | GOT1, ERLIN1 ENSOCUG00000014801, ENSOCUG00000001375 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laghouaouta, H.; Sosa-Madrid, B.S.; Zubiri-Gaitán, A.; Hernández, P.; Blasco, A. Novel Genomic Regions Associated with Intramuscular Fatty Acid Composition in Rabbits. Animals 2020, 10, 2090. https://doi.org/10.3390/ani10112090
Laghouaouta H, Sosa-Madrid BS, Zubiri-Gaitán A, Hernández P, Blasco A. Novel Genomic Regions Associated with Intramuscular Fatty Acid Composition in Rabbits. Animals. 2020; 10(11):2090. https://doi.org/10.3390/ani10112090
Chicago/Turabian StyleLaghouaouta, Houda, Bolívar Samuel Sosa-Madrid, Agostina Zubiri-Gaitán, Pilar Hernández, and Agustín Blasco. 2020. "Novel Genomic Regions Associated with Intramuscular Fatty Acid Composition in Rabbits" Animals 10, no. 11: 2090. https://doi.org/10.3390/ani10112090