The Identification of Runs of Homozygosity Gives a Focus on the Genetic Diversity and Adaptation of the “Charolais de Cuba” Cattle

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Used and Marker Quality Controls
2.2. Detection of Runs of Homozygosity (ROH)
2.3. Estimation of Genomic Inbreeding
2.4. Estimation of Effective Population Size
2.5. Candidate Regions Responsible of the CHCU Adaptation to Tropical Conditions
3. Results
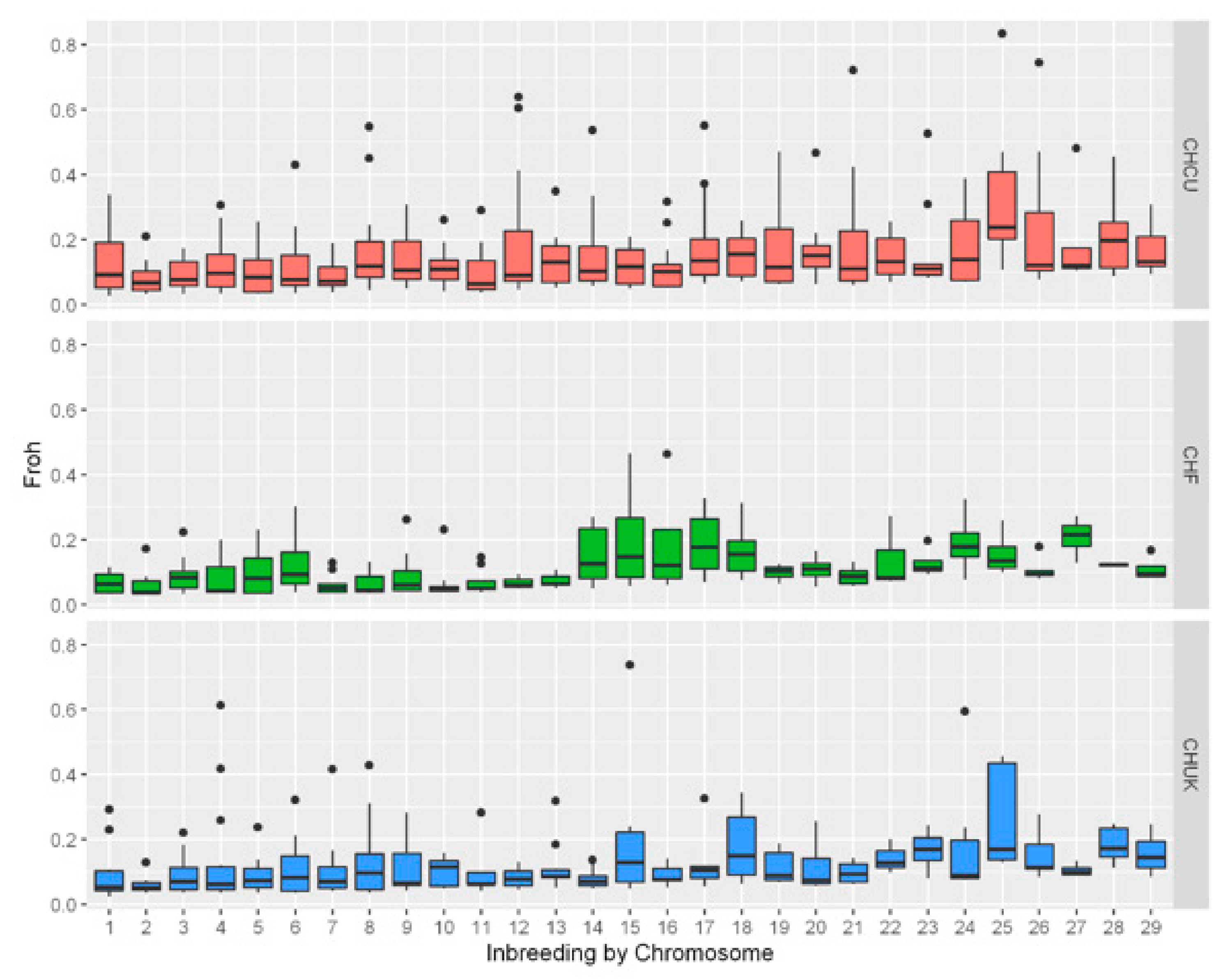
3.1. Estimation of Runs of Homozygosity (ROH) and Genomic Inbreeding
3.2. Estimation of Effective Populations Size
3.3. Candidate Regions Responsible of the CHCU Adaptation to Tropical Conditions
4. Discussion
4.1. Limitations and Trade-Offs of the Analysis Based on Genotype Datasets and Small Sample Sizes
4.2. Estimation of Runs of Homozygosity (ROHs) and Genomic Inbreeding
4.3. Estimation of Effective Populations Size
4.4. Candidate Regions Responsible of the CHCU Adaptation to Tropical Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López, D.; Miriam, R.; Díaz, J.; Menéndez, A. Introducción y desarrollo del Charolais en Cuba. In En El Charolais Cubano; Editorial Científico-Técnica: La Habana, Cuba, 1977; pp. 13–17. (In Spanish) [Google Scholar]
- Wright, S. Coefficients of Inbreeding and Relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–360. [Google Scholar] [CrossRef]
- Silió, L.; Rodríguez, M.C.; Fernández, A.; Barragán, C.; Benítez, R.; Óvilo, C.; Fernández, A.I. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. J. Anim. Breed. Genet. 2013, 130, 349–360. [Google Scholar] [CrossRef]
- Fernández, A.; Angel Toro, M.; López-Fanjul, C. The effect of inbreeding on the redistribution of genetic variance of fecundity and viability in Tribolium castaneum. Heredity 1995, 75, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; De Jong, G.; Hiemstra, S.J.; Windig, J.J. Inbreeding depression due to recent and ancient inbreeding in Dutch Holstein-Friesian dairy cattle. Genet. Sel. Evol. 2019, 51, 54. [Google Scholar] [CrossRef] [Green Version]
- Curik, I.; Ferenčakovic, M.; Sölkner, J. Genomic dissection of inbreeding depression: A gate to new opportunities. Rev. Bras. Zootec. 2017, 46, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Goszczynski, D.; Molina, A.; Terán, E.; Morales-Durand, H.; Ross, P.; Cheng, H.; Giovambattista, G.; Demyda-Peyrás, S. Runs of homozygosity in a selected cattle population with extremely inbred bulls: Descriptive and functional analyses revealed highly variable patterns. PLoS ONE 2018, 13, e0200069. [Google Scholar] [CrossRef] [Green Version]
- Nothnagel, M.; Tehua, T.L.; Kayser, M.; Krawczak, M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010, 19, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Corbin, L.J.; Liu, A.Y.H.; Bishop, S.C.; Woolliams, J.A. Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 2012, 129, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, T.J. Estimating Effective Population Size from Genetic Data: The Past, Present, and the Future. Ph.D. Thesis, Imperial College London, London, UK, January 2017. [Google Scholar]
- Xu, L.; Zhu, B.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of linkage disequilibrium, effective population size and haplotype block structure in Chinese cattle. Animals 2019, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconer, D.S.; Mackay, T.F.C. Effective populations size. In Introduction into Quantitative Gentics, 3rd ed.; Longman Scientific & Technical: Harlow, UK, 1989; pp. 70–76. [Google Scholar]
- Saura, M.; Tenesa, A.; Woolliams, J.A.; Fernández, A.; Villanueva, B. Evaluation of the linkage-disequilibrium method for the estimation of effective population size when generations overlap: An empirical case. BMC Genom. 2015, 16, 922. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Hill, W.G. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Valera, Y.; Renand, G.; Naves, M.; Fonseca-Jiménez, Y.; Moreno-Probance, T.I.; Ramos-Onsins, S.; Rocha, D.; Ramayo-Caldas, Y. Genetic diversity and selection signatures of the beef “Charolais de Cuba” breed. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and Characterization of a High Density SNP Genotyping Assay for Cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [Green Version]
- Gautier, M.; Laloe, D.; Moazami-Goudarzi, K. Insights into the Genetic History of French Cattle from Dense SNP Data on 47 Worldwide Breeds. PLoS ONE 2010, 5, e13038. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. Plink: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben, J.S.; Boussaha, M.; Thamri, N.; Mnara, S.; Rebours, E.; Rocha, D. Linkage disequilibrium and past effective population size in native Tunisian cattle. Genet. Mol. Biol. 2019, 42, 52–61. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: An R Package to Detect Runs of Homozygosity and Heterozygosity in Diploid Genomes. Sample Data 2018. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 25 January 2020).
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; MacCiotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- Hayes, B.J.; Visscher, P.M.; McPartlan, H.C.; Goddard, M.E. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Ohta, T.; Kimura, M. Linkage disequilibrium between two segregating nucleotide sites under the steady flux of mutations in a finite population. Genetics 1971, 68, 571–580. [Google Scholar]
- Tenesa, A.; Navarro, P.; Hayes, B.J.; Duffy, D.L.; Clarke, G.M.; Goddard, M.E.; Visscher, P.M. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007, 17, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Purfield, C.D.; Berry, P.D.; McParland, S.; Bradley, G.D.; Burgess, H.A.; Martinez, S.; Reiner, O. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 58, 70. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Buxadera, A. Variación genética de la composición de la canal en la raza Charolais. Ph.D. Thesis, Instituto Superior de Ciencias Agropecuarias de La Habana, La Habana, Cuba, November 1978; pp. 2–6. (In Spanish). [Google Scholar]
- Felius, M.; Beerling, M.L.; Buchanan, D.S.; Theunissen, B.; Koolmees, P.A.; Lenstra, J.A. On the history of cattle genetic resources. Diversity 2014, 6, 705–750. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Thomas, P. Panther pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [PubMed]
- Watterson, G. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256. [Google Scholar] [CrossRef]
- Weigel, K.A. Controlling Inbreeding in Modern Breeding Programs. J. Dairy Sci. 2001, 84, E177–E184. [Google Scholar] [CrossRef]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- François, L.; Wijnrocx, K.; Colinet, F.G.; Gengler, N.; Hulsegge, B.; Windig, J.J.; Buys, N.; Janssens, S. Genomics of a revived breed: Case study of the Belgian campine cattle. PLoS ONE 2017, 12, e0175916. [Google Scholar] [CrossRef] [Green Version]
- Msalya, G.; Kim, E.S.; Laisser, E.L.K.K.; Kipanyula, M.J.; Karimuribo, E.D.; Kusiluka, L.J.M.M.; Chenyambuga, S.W.; Rothschild, M.F. Determination of genetic structure and signatures of selection in three strains of Tanzania Shorthorn Zebu, Boran and Friesian cattle by genome-wide SNP analyses. PLoS ONE 2017, 12, e171088. [Google Scholar] [CrossRef] [Green Version]
- Solé, M.; Gori, A.; Faux, P.; Bertrand, A.; Farnir, F.; Gautier, M.; Druet, T. Age-based partitioning of individual genomic inbreeding levels in Belgian Blue cattle. Genet. Sel. Evol. 2017, 49, 92. [Google Scholar] [CrossRef] [Green Version]
- Cortés, O.; Eusebi, P.; Dunner, S.; Sevane, N.; Cañón, J. Comparison of diversity parameters from SNP, microsatellites and pedigree records in the Lidia cattle breed. Livestig. Sci. 2019, 219, 80–85. [Google Scholar] [CrossRef]
- Toro-Ospina, M.A.; da Silva-Faria, R.A.; Vercesi-Filho, A.E.; dos Santos-Goncalves, J.N.C.; Zerlotti-Mercadante, M.E.; Abdallah-Curi, R.; de Vasconcelos-Silva, J.A.I. Genome-wide identification of runs of homozygosity islands in the Gyr breed (Bos indicus). Reprod. Domest. Anim. 2020, 55, 333–342. [Google Scholar] [CrossRef]
- Toro-Ospina, A.M.; Maiorano, A.M.; Curi, R.A.; Pereira, G.L.; Zerlotti-Mercadante, M.E.; dos Santos Gonçalves Cyrillo, J.N.; Aspilcueta-Borquis, R.R.; Josineudson, J.A.I.I. Linkage disequilibrium and effective population size in Gir cattle selected for yearling weight. Reprod. Domest. Anim. 2019, 54, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Šidlová, V.; Kasarda, R.; Moravčíková, N.; Trakovická, A.; Curik, I.; Ferenčaković, M. Genomic variability among cattle populations based on runs of homozygosity. Poljoprivreda 2015, 21, 44–47. [Google Scholar] [CrossRef]
- Kasarda, R.; Kadlečík, O.; Trakovická, A.; Moravčíková, N.; Biology, B. Genomic and pedigree-based inbreeding in Slovak Spotted Cattle. AGROFOR Int. J. 2019, 4, 102–110. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Zabek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livestig. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Lu, D.; Sargolzaei, M.; Kelly, M.; Li, C.; Voort, G.V.; Wang, Z.; Plastow, G.; Moore, S.; Miller, S.P. Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front. Genet. 2012, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Cheong, H.S.; Shin, H.D.; Lee, S.S.; Roh, H.J.; Jeon, D.Y.; Cho, C.Y. Genetic diversity and divergence among Korean cattle breeds assessed using a BovineHD single-nucleotide polymorphism chip. Asian-Australas. J. Anim. Sci. 2018, 31, 1691–1699. [Google Scholar] [CrossRef]
- Aguilar, R.; Bu, A.; Dresdner, J.; Fernández, P.; González, A.; Polanco, C.; Tansini, R. Capítulo 1. Antecedentes de la ganadería vacuna en Cuba. In La ganadería en Cuba.: Desempeño y Desafíos; ASDI : INIE : Udelar. FCS-DE: Montevideo, Uruguay, 2004; p. 15. ISBN 959-7160-16-1. [Google Scholar]
- Bouquet, A.; Venot, E.; Laloë, D.; Forabosco, F.; Fogh, A.; Pabiou, T.; Moore, K.; Eriksson, J.; Renand, G.; Phocas, F. Genetic structure of the European Charolais and Limousin cattle metapopulations using pedigree analyses. J. Anim. Sci. 2011, 89, 1719–1730. [Google Scholar] [CrossRef]
- Ribas, M. Gene frequencies in the blood group systems of the Cuban Charolais. Ann. Génét. Sél. Anim. 1981, 13, 293–300. [Google Scholar]
- FAO. Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans: Management of Small Populations at Risk; FAO: Rome, Italy, 1998; Available online: http://www.fao.org/3/a-w9361e.pdf (accessed on 20 June 2019).
- Pavlidis, P.; Jensen, J.D.; Stephan, W.; Stamatakis, A. A critical assessment of storytelling: Gene ontology categories and the importance of validating genomic scans. Mol. Biol. Evol. 2012, 29, 3237–3248. [Google Scholar] [CrossRef] [Green Version]
- Direito, I.; Madeira, A.; Brito, M.A.; Soveral, G. Aquaporin-5: From structure to function and dysfunction in cancer. Cell. Mol. Life Sci. 2016, 73, 1623–1640. [Google Scholar] [CrossRef]
- Rout, M.; Panigrahi, S.; Pradhan, S.; Swain, K. Genetic basis of heat tolarance in cattle. Pharma Innov. J. 2018, 7, 183–185. [Google Scholar]
- Collier, R.J.; Collier, J.L.; Rhoads, R.P.; Baumgard, L.H. Invited review: Genes involved in the bovine heat stress response. J. Dairy Sci. 2008, 91, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large Potentials of Small Heat Shock Proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, L.; Gonzatti, M.I.; Thevenon, S.; Chantal, I.; Pinto, J.; Berthier, D.; Aso, P.M.; Gautier, M. A quasi-exclusive European ancestry in the Senepol tropical cattle breed highlights the importance of the slick locus in tropical adaptation. PLoS ONE 2012, 7, e36133. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Bickhart, D.M.; Landaeta-hernandez, A.J.; Utsunomiya, Y.T.; Pagan, M.; Jimenez, E.; Hansen, P.J.; Dikmen, S.; Schroeder, S.G.; Kim, E.; et al. Convergent Evolution of Slick Coat in Cattle through Truncation Mutations in the Prolactin Receptor. Front. Genet. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Davila, K.M.S.; Howell, A.; Nunez, A.; Orelien, A.; Roe, V.; Rodriguez, E.; Dikmen, S.; Mateescu, R.G. Genome-wide association study identifies variants associated with hair length in Brangus cattle. Anim. Genet. 2020, 1–4. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Tu, W.-L.; Chen, C.-J.; Chan, H.-L.; Chen, C.-F.; Chen, H.-H.; Tang, P.-C.; Lee, Y.-P.; Chen, S.-E.; Huang, S.-Y. Functional genomics study of acute heat stress response in the small yellow follicles of layer-type chickens. Sci. Rep. 2018, 8, 1320. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.T.; Kachman, S.D.; Snelling, W.M.; Pollak, E.J.; Ciobanu, D.C.; Kuehn, L.A.; Spangler, M.L. Beef cattle body temperature during climatic stress: A genome-wide association study. Int. J. Biometeorol. 2014, 58, 1665–1672. [Google Scholar] [CrossRef] [Green Version]
- Flori, L.; Moazami, K.; Véronique, G.; Araba, A.; Boujenane, I.; Boushaba, N.; Casabianca, F.; Casu, S.; Ciampolini, R.; Coeur, A.; et al. A genomic map of climate adaptation in Mediterranean cattle breeds. Mol. Ecol. 2019, 28, 1009–1029. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Breeds | 4–8 Mb FROH (±SD) | 8–16 Mb FROH (±SD) | >16 Mb FROH (±SD) |
|---|---|---|---|
| CHF | 0.034 (0.012) | 0.017 (0.011) | 0.007 (0.007) |
| CHCU | 0.057 (0.025) | 0.037 (0.022) | 0.016 (0.016) |
| CHUK | 0.040 (0.022) | 0.023 (0.019) | 0.011 (0.014) |
| Interval (Mb) | CHF | CHCU | CHUK | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Number | Freq (%) | MNROH (Mb) | Number | Freq (%) | MNROH (Mb) | Number | Freq (%) | MNROH (Mb) | |
| 4–8 | 158 | 71.5 | 5.35 | 354 | 58.3 | 5.549 | 193 | 65.6 | 5.598 |
| 8–16 | 47 | 21.3 | 11.072 | 182 | 29.9 | 11.24 | 73 | 24.9 | 10.633 |
| >16 | 16 | 7.2 | 22.195 | 72 | 11.8 | 23.634 | 28 | 9.5 | 27.377 |
| Populations | Class Mb | NROH | MNROH (Mb) | ±SD | CV % | IC −95% | IC +95% |
|---|---|---|---|---|---|---|---|
| CHF | 4–8 | 158 | 5.34 | 1.08 | 20.28 | 5.18 | 5.52 |
| 8–16 | 47 | 11.07 | 2.27 | 20.49 | 10.40 | 11.73 | |
| >16 | 16 | 22.19 | 5.65 | 25.49 | 19.18 | 25.21 | |
| CHCU | 4–8 | 354 | 5.54 | 1.14 | 20.69 | 5.42 | 5.67 |
| 8–16 | 182 | 11.24 | 2.19 | 19.51 | 10.91 | 11.56 | |
| >16 | 72 | 23.63 | 9.35 | 39.58 | 21.43 | 25.83 | |
| CHUK | 4–8 | 193 | 5.60 | 1.14 | 20.37 | 5.44 | 5.76 |
| 8–16 | 73 | 10.63 | 2.07 | 19.47 | 10.15 | 11.11 | |
| >16 | 28 | 27.37 | 13.22 | 48.29 | 22.25 | 32.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Valera, Y.; Rocha, D.; Naves, M.; Renand, G.; Pérez-Pineda, E.; Ramayo-Caldas, Y.; Ramos-Onsins, S.E. The Identification of Runs of Homozygosity Gives a Focus on the Genetic Diversity and Adaptation of the “Charolais de Cuba” Cattle. Animals 2020, 10, 2233. https://doi.org/10.3390/ani10122233
Rodríguez-Valera Y, Rocha D, Naves M, Renand G, Pérez-Pineda E, Ramayo-Caldas Y, Ramos-Onsins SE. The Identification of Runs of Homozygosity Gives a Focus on the Genetic Diversity and Adaptation of the “Charolais de Cuba” Cattle. Animals. 2020; 10(12):2233. https://doi.org/10.3390/ani10122233
Chicago/Turabian StyleRodríguez-Valera, Yoel, Dominique Rocha, Michel Naves, Gilles Renand, Eliecer Pérez-Pineda, Yuliaxis Ramayo-Caldas, and Sebastian E. Ramos-Onsins. 2020. "The Identification of Runs of Homozygosity Gives a Focus on the Genetic Diversity and Adaptation of the “Charolais de Cuba” Cattle" Animals 10, no. 12: 2233. https://doi.org/10.3390/ani10122233